Missing clinical trial data
by Richard Lehman and Elizabeth Loder
British Medical Journal 2012 344:d8158.
Clinical medicine involves making decisions under uncertainty. Clinical research aims to reduce this uncertainty, usually by performing experiments on groups of people who consent to run the risks of such trials in the belief that the resulting knowledge will benefit others. Most clinicians assume that the complex regulatory systems that govern human research ensure that this knowledge is relevant, reliable, and properly disseminated. It generally comes as a shock to clinicians, and certainly to the public, to learn that this is far from the case. The linked cluster of papers on unpublished evidence should reinforce this sense of shock. These articles confirm the fact that a large proportion of evidence from human trials is unreported, and much of what is reported is done so inadequately. We are not dealing here with trial design, hidden bias, or problems of data analysis — we are talking simply about the absence of the data. And this is no academic matter, because missing data about harm in trials can harm patients, and incomplete data about benefit can lead to futile costs to health systems. Moreover, researchers or others who deliberately conceal trial results have breached their ethical duty to trial participants…The main challenge is to ensure better systems for the future. Because “the optimal systematic review would have complete information about every trial — the full protocol, final study report, raw dataset, and any journal publications and regulatory submissions,” a prospective system of research governance should insist on nothing less. This may require the global organisation of a suitable shared database for all raw data from human trials — an obvious next step for the World Health Organization after its excellent work on the International Clinical Trials Registry Platform Search Portal. Concealment of data should be regarded as the serious ethical breach that it is, and clinical researchers who fail to disclose data should be subject to disciplinary action by professional organisations. This may achieve quicker results than legislation in individual countries, although this is also desirable. These changes have been long called for, and delay has already caused harm. The evidence we publish shows that the current situation is a disservice to research participants, patients, health systems, and the whole endeavour of clinical medicine.
Compliance with mandatory reporting of clinical trial results on ClinicalTrials.gov: cross sectional study
by Andrew P Prayle, Matthew N Hurley, and Alan R Smyth
British Medical Journal 2012 344:d7373.
[full text on-line]
Objective: To examine compliance with mandatory reporting of summary clinical trial results (within one year of completion of trial) on ClinicalTrials.gov for studies that fall under the recent Food and Drug Administration Amendments Act (FDAAA) legislation.
Design: Registry based study of clinical trial summaries.
Data sources: ClinicalTrials.gov, searched on 19 January 2011, with cross referencing with Drugs@FDA to determine for which trials mandatory reporting was required within one year.
Selection criteria: Studies registered on ClinicalTrials.gov with US sites which completed between 1 January and 31 December 2009.
Main outcome measure: Proportion of trials for which results had been reported.
Results: The ClinicalTrials.gov registry contained 83 579 entries for interventional trials, of which 5642 were completed within the timescale of interest. We identified trials as falling within the mandatory reporting rules if they were covered by the FDAAA (trials of a drug, device, or biological agent, which have at least one US site, and are of phase II or later) and if they investigated a drug that already had approval from the Food and Drug Administration. Of these, 163/738 (22%) had reported results within one year of completion of the trial compared with 76/727 (10%) trials that were not subject to mandatory reporting (95% confidence interval for the difference in proportions 7.8% to 15.5%; χ² test, P<0.001. Later phase trials were more likely to report results (P<0.001, as were industry funded trials (P<0.001).
Conclusion: Most trials subject to mandatory reporting did not report results within a year of completion.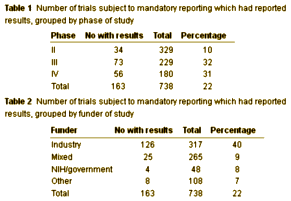
I’m not well enough acquainted with H.R. 3580, the Food and Drug Administration Amendments Act of 2007, or for that matter, H.R. 6272, the proposed TEST Act of 2012, to know what they have to say about data format or enforcement, and I’m too constitutionally averse to government-ese to be the one to parse out the details. But I think in the case of psychiatry, I’d like to see the availability of raw data become a prerequisite for publishing in a peer reviewed journal or applying for FDA Approval. Why not? There’s no reason not to. If you can write a paper or apply for approval, you have the data already. It doesn’t need prep time. In fact the whole point is un-prep, to have unprocessed data available for vetting.
 can be accessed, people will do what’s needed to check the facts, particularly when the kind of gross conflicts of interest we’ve lived with for a quarter century are in play. The drug companies complain that they already have to jump through too many hoops and prominent people like Dr. Insel of the NIMH [Experimental Medicine] and the KOL Klan that met at the Pipeline Summit earlier this year seem to buy their lament, calling for loosened regulations and streamlined processes. Given the track record of the recent era, I’d say bring on the hoops! [at least the ones that matter]…
can be accessed, people will do what’s needed to check the facts, particularly when the kind of gross conflicts of interest we’ve lived with for a quarter century are in play. The drug companies complain that they already have to jump through too many hoops and prominent people like Dr. Insel of the NIMH [Experimental Medicine] and the KOL Klan that met at the Pipeline Summit earlier this year seem to buy their lament, calling for loosened regulations and streamlined processes. Given the track record of the recent era, I’d say bring on the hoops! [at least the ones that matter]…
You are lucky to be leaving the game soon, I have summed up what psychiatry basically is and will be hereon as the 3 A’s, Triple A if it fits better:
Addiction/controlled sub seeking, Antisocial/forensic focusing, and Acute/chronic care limiting to hospitals and community mental health clinics.
You see, the stupidity and short sightedness of all these narcicisstic, greedy narrow minded “leaders” have lead us to be pigeon holed into these three areas, and while the first two are “legitimate” subspecialties that have some application in our profession, they have metastisized into the erroneous assumption that all psychiatrists do these things. And my beef with the third part, acute/chronic care, is just letting the nonpsychiatrists cherry pick patients who have opportunity to do good psychotherapy work and be guided to remission to not be accessed by psychiatrists. Well, that is nice if you know what you are doing with the alleged cherries of the bunch, but even therapists now are quick to label adjustment disorder type presentations to some type of prescriber because, hey, therapists don’t have time to do real therapy work these days, at least a fair number I have witnessed in my travels the past 7 or more years.
So, psychiatry is now the AAA group. Really will drum up residency options for programs all across the land. Well, only those who get the 329 studies, eh?