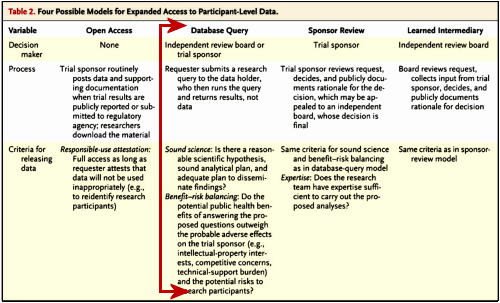
Harvard CimsonBy Emma C. CobbOct 30, 2013Researchers from Harvard, together with members of a group created by the Multi-Regional Clinical Trials Center, released a report last week that proposed ways to expand access to clinical trial data. The report–published online in The New England Journal of Medicine–outlined the possible benefits and risks, ethical and legal issues, and logistical questions associated with expanding access to participant-level data, which have, in the past, been considered confidential by the Food and Drug Administration, which regulates clinical trials of drugs conducted in the US.
Addressing concerns about sponsor-supported results, the authors stated that expanded access to clinical data could allow for a greater scientific innovation and a more accurate representation of a product’s safeness and effectiveness. “Our experiences with Vioxx, Avandia, and other widely prescribed drugs that were revealed to have serious safety risks show how important it is to give independent scientists access to clinical trial data,” said Michelle M. Mello, a professor at Harvard School of Public Health and lead author of the report, in a press release. While Mello emphasized the importance of providing independent scientists with access to clinical trial data, she also cautioned against the risk of revealing participants’ identities and providing too much information, which might strain trial sponsors. “The question is, how can we achieve the powerful public health benefits of data sharing while protecting research participants’ privacy, avoiding ‘junk science,’ and minimizing burdens on trial sponsors?” Mello said in the release.
The report also discussed current initiatives to expand data access. For instance, the European Medicines Agency recently announced that it will start providing participant-level data that is submitted for regulatory purposes. The authors recommended that new regulations targeted at expanding data sharing should be applied to trials of all approved prescription drugs, medical devices, and biologics in a way that preserves research participant privacy and treats sponsors and researchers “evenhandedly.” Although the report does not come to a conclusion about how access to data should be expanded, it emphasizes that the expanded access to data is a question of how the transparency should be achieved, not whether or not it will.
hat tip to pharmagossip…
-
Data Sharing: This is a PHARMA/NIMH creation that reframes the whole game. As soon as you’ve been engaged with the notion of data sharing, you’ve drunk the kool-aid. In the service of furthering the scientific mission, the benevolent pharmaceutical industry is sharing its owned and paid for data for the good of mankind.
-
Patient/Subject/Participant Confidentiality: The right to privacy in just part of living in a free sociaty. If the Clinical Trial Sponsor is so concerned about confidentiality, why are they the ones involved in this whole debate? If things worked right, they shouldn’t know who the subjects are themselves. These studies are done by CRO [Clinical Research Organizations] or multi-centered academic sites. The responsibility for confidentiality is with the person or persons who actually has contact with the subject. So, for example, GSK ought to be as in the dark as anyone else about the subject identity. If privacy is that important, the information sent to them ought to be as anonymized as to anyone else. I’m not sure subject confidentiality and patient confidentiality are the same things, but that’s not a widely held view so I’ll keep it to myself.
-
Proxies: If the Sponsor publishes the data in an academic journal, the article is merely a proxy for the data [proxies…], not the data itself. Eveything to the right of the red line makes the mistake best expressed by the Zen story about confusing the finger for the Moon or the Existentialist credo, existence precedes essence or the line in Jerry McGuire, Show me the money.
-
Cheating: The only reason for the data to be held behind a firewall is to distort it. It’s not intellectual property, it’s numbers that are supposedly already informing the published work. There are too many examples of distortion to even suggest this isn’t true. I don’t care a whit about sharing data. I want to monitor what they’re doing because without oversight, they cheat. They’ve cheated a lot – maybe most of the time. This is not poker or any other kind of game. This is medicine. If they can’t show all their cards, they shouldn’t be here.
If you’re tempted to believe that this article gets to the heart of the conflict, go listen to Neal Parker one more time [start @ 19:00].
Source InformationFrom the Department of Health Policy and Management, Harvard School of Public Health (M.M.M.), the Department of Medicine, Brigham and Women’s Hospital and Harvard Medical School (B.E.B.), and Ropes and Gray (M.B.) — all in Boston; Multi-Regional Clinical Trials Center at Harvard (M.W., B.E.B., M.B.) and Harvard Law School (M.B.) — both in Cambridge, MA; Pharmaceutical Research and Manufacturers of America, Washington, DC (J.K.F.); Stonington Heights Consulting, Briarcliff Manor, NY (M.W.); Core Risks, Ardmore, PA (M.W.); and Teden Consulting, South Orange, NJ (P.T.).
You do realize Harvard is the citadel of biopsychiatry?
I used to know it, but I know it more now than I used to know it!
I actually agree with you that subjects are not the same as patients. But even assuming the two are the same, in the future, why not get consent? I mean, a regulatory body could require that pharmaceutical companies get consent from subjects for making their anonymized data available to the world. They get consent to put potential poisons into subjects’ bodies. This could just be one more page on the consent forms.
And if, say, the FDA required that any trial submitted for drug approval needed to have these consents, or the drug would not be considered for approval, I think the pharmaceutical companies would have to comply, and could no longer object to release of information to the public on the basis of maintaining patient privacy.
Of course, even if this weren’t a fantasy, it would do nothing for data from trials that have already taken place, and it would do nothing about the issue of drug companies believing, or posturing, that their data is proprietary.
PsychPractice
“I actually agree with you that subjects are not the same as patients. But even assuming the two are the same, in the future, why not get consent? I mean, a regulatory body could require that pharmaceutical companies get consent from subjects for making their anonymized data available to the world. They get consent to put potential poisons into subjects’ bodies. This could just be one more page on the consent forms.”
The only think I can think of to say to that is a simple “Amen.” Why not?
Do you know anything about potential changes in legislature for the FDA? As I understand it, right now, the FDA doesn’t have much power, but if there were tighter requirements, and these were enforced, a lot could be accomplished.