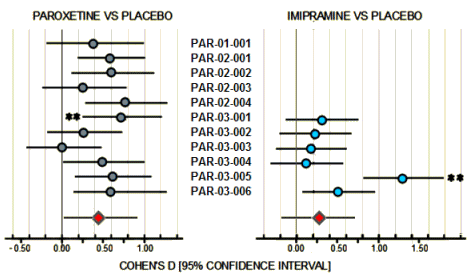
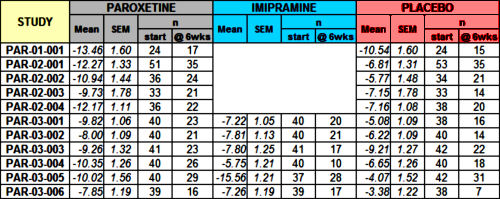
Just scanning the figure, it looks okay. 7/11 of the Paroxetine trials are in the moderate range and are statistically significant. By FDA standards, this passes the efficacy requirements with flying colors. It looks to leave Imipramine in the dust with only 2/6 studies in the moderate range and statistically significant. However, the Paroxetine/Imipramine difference is an illusion. The only statistically significant differences in those 6 identical studies are the two marked with asterisks, one favoring each drug. And the weighted composites [in red] are likewise not significantly different. The unmentioned 6 trials were either uncontrolled or failed trials with recruitment/drop-out problems. The values shown above for the 5/6 of the trials against Imipramine are from only 4 weeks because by the time the planned 6 week period came around, there had been too many drop-outs to make valid calculations. The overall dropout rate by 6 weeks for the 11 trials was ~50%.
Finally, if you look at the breadth of the 95% Confidence Intervals in the figure, there was a lot of variability and many subjects that would fall out of the clinically effective therapeutic range.
These problems of drop-outs, missing data, wide variability among the subjects in a given study or between separate studies, not really knowing if the patients are help-seeking or paid recruits, etc. are part and parcel of most clinical trials whether run in an academic center, by a large Clinical Research Organization, or by a small center like those in Carl Elliot’s recent piece [under some of the rocks…]. Human beings don’t have uniform illnesses, don’t have the genetic uniformity of cloned white mice, aren’t confined to wire cages on a strictly controlled diet. So even in the most pristine and unbiased of trials there’s an intrinsic heterogeneity that transcends the clean demographic tables found in the published articles. And in psychiatry, the subjectivity of the symptoms just adds to the confusion.
The main charge of the FDA is safety, adverse events, and I didn’t look at that part of this NDA [maybe another time]. Proof of efficacy was added in 1962 by the Kefauver-Harris Amendment to keep the ineffective patent medicines out of our pharmacopoeia. The efficacy criteria are minimal – two well conducted studies demonstrated efficacy. This Paroxetine submission easily achieves that requirement. It seems that the drug, Paroxetine, does have antidepressant properties, but that’s all it means. Leaving aside the crucial issue of harms for the moment, that’s what the FDA is for – certifying that the drug does what its sponsor says it does, that it’s not snake oil or some inert substance of no therapeutic value. How well it does it or how often or in whom or when indicated are another matter.
When I found this disc, I was initially curious because it came from several decades ago. It would be surprising to find single site trials these days. It’s hard to recruit the number of subjects needed to show differences [called power] at a single site. Nowadays, the trials use multiple sites to achieve the necessary power [and they’re a lot faster]. This study is from a time before the meteoric rise of the Clinical Research Organizations, so these trials were at the smaller clinical research centers of the time or academic institutions. I expected [and found] that the NDA would show why Clinical Trials are really not some bottom line knowledge [beyond…], just a piece of data among the many other things that go into a clinical recommendation. Talking about them, people like to use the word confounders. and RTCs always have confounders like in this study.
 There are two always-available criticisms of physicians’ decisions: "That’s just what you think! What’s your evidence base?" and "You’re just following a guideline by rote and not seeing the person in front of you!" At one time or another, each of those negative epithets might well be accurate – sometimes both apply. But somewhere in recent times, the battle cry of evidence based medicine has shifted the balance and fostered the notion that the guidelines derived from groups dictate the best course for an individual case
There are two always-available criticisms of physicians’ decisions: "That’s just what you think! What’s your evidence base?" and "You’re just following a guideline by rote and not seeing the person in front of you!" At one time or another, each of those negative epithets might well be accurate – sometimes both apply. But somewhere in recent times, the battle cry of evidence based medicine has shifted the balance and fostered the notion that the guidelines derived from groups dictate the best course for an individual case  – implying a uniformity among people and a strict objectivity to medical care that is illusory. Randomized Clinical Trials, Rating Scales, Statistical Significance, FDA Approval, Expert Opinions, and all the other ways we try to extract objective markers from subjective phenomena are vital tools in the medical toolbox, but only that – tools…
– implying a uniformity among people and a strict objectivity to medical care that is illusory. Randomized Clinical Trials, Rating Scales, Statistical Significance, FDA Approval, Expert Opinions, and all the other ways we try to extract objective markers from subjective phenomena are vital tools in the medical toolbox, but only that – tools…
Along the same lines:
http://www.medrants.com/archives/7842
I am also concerned by the time frame of six weeks. Past post have shown the effectiveness of these medications to decline at the 12 week mark and bottom out at 16 weeks. Often when looking at the time function of medications we see the original trials structured so as to stop when there is a positive outcome and not carry through long enough to show the negative side of the product being tested.
This has all become part of the pharma game.
Steve Lucas
Hot off the web:
http://www.forbes.com/sites/larryhusten/2014/08/18/an-old-study-fuels-debate-over-blood-pressure-guidelines/
Steve Lucas
In response to Steve’s 2nd comment, I wanted to alert people that if you are diagnosed with high blood pressure, you might want to ask for a 24 hour blood pressure monitor to confirm the diagnosis. Fortunately, I did that and mine turned out to be normal as the high office readings were due to white coat hypertension.
I know this may be considered an off topic comment but big pharma doesn’t alert patients to do this.
Actually, it’s not going in my library because I don’t have access to most of them.