
The European Medicines Agency’s Data Transparency policy has made many twists and turns along the way – so many that I summarized them with a timeline last October to serve as a placemarker for the day when the implementation was released. That Timeline is available for review. Tom Jefferson has reported on their recent 2+ hour progress update Webinar in a couple of BMJ blogs – mercifully short and very clear. By my read, the news is good:
BMJ Blogsby Tom Jefferson30 Jun, 15
BMJ Blogsby Tom Jefferson30 Jun, 15
For twenty five years, industry funded RCTs flooded the psychiatric literature, many grossly distorted – creating an illusion of safety and efficacy that produced billions of dollars in revenue for the pharmaceutical industry. There was no way to check those reports, because the actual data was a secret kept by the companies who financed the trials and sold the drugs. And so there was a major campaign to get that raw data into the public domain where it could be independently analyzed. The EMA lead the way vowing to make it available, in fits and starts, as outlined in the Timeline.
But be careful what you ask for. You might just get it. There are literally thousands of pages with each RCT. I’ve spent the last year and a half working on just one trial and it’s a maddening exercise, bouncing around in these documents looking for elusive needles among the hay. So how this data is presented and cataloged is as important as its availability. Tom’s blogs are about how the EMA is going to implement Phase I of their roll-out [the operative saying being, "the devil’s in the details"]. Here’s my monotonous graphic, updated to fit the new EMA proposals:
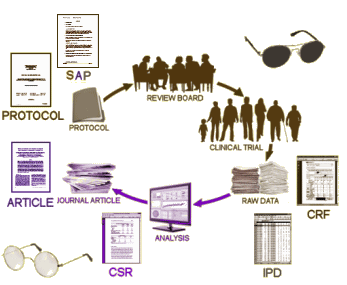
There are many ways to subtly distort to results of a clinical trial without committing actual fraud. The consensus is that having the raw data for a complete reanalysis is the only preventive. What are the essential pieces necessary? First, the investigator needs to specify in detail exactly how the study will be conducted; how the data will be collected; how it will be analyzed; and what criteria will be used to judge the results. All of this needs to be specified in advance. Anyone who has ever done a study knows that once you have the results, you can massage the data and make it say all sorts of things. So the PROTOCOL and the SAP [Statistical Analysis Plan] need to be prepared before the study begins [a priori]:
-
from a general law to a particular instance;
valid independently of observation. -
existing in the mind independent of experience.
-
conceived beforehand.
a pri·o·ri [ä′ prë-ôr′ë]
adj.
Phase I plans to make the CSR [Clinical Study Report] available promptly after any regulatory action by the EMA. The reason I say it "looks good" is that they specify what’s in the CSR:
Phase 1 covers the release of clinical overviews [Module 2.5 in regulatory speak], clinical summaries [Module 2.7], and the all important clinical study reports [CSRs]. CSRs will include the main body [a sort of IMRAD style document], the trial protocol with its dated amendments, blank (or sample) case report forms [CRFs], and the statistical analysis plan or SAP [appendices]. See this exploratory review for a description of each cited document.
The value of these documents available at the click of a button to a researcher is difficult to overplay. For example, Module 2.5 contains a complete description of the manufacturer’s trial programme. For those of us who laboured for six months to reconstruct trial programmes, cross-referencing fragments of information from disparate sources, this is the honey pot served on a plate. CSRs are the most detailed accounts of the setting up, conduct, and analyses of trials on pharmaceuticals by their sponsors.
The original offerings as the companies began to realize that they were going to have to go along with transparency was to make the CSRs available. It’s an extensive write-up on the study in narrative form with the results. The problem was that what was in those reports wasn’t specified. And in practice, the distortions and obfuscations that appeared in the articles could be just as easily put into the CSR. With the "trial protocol with its dated amendments, blank [or sample] case report forms [CRFs], and the statistical analysis plan or SAP [appendices]" specified, one has everything needed to check to see if the a priori defined study is the one they are reporting. This information will be available within 60 days of a drug approval and they have a reasonable process to manage redactions. The only things missing are the raw CRFs and the IPDs [tables of individual results]. That will be in Phase II. The CRFs would be required for a thorough vetting of the Adverse Events and the IPDs to actually repeat the statistical analyses. But otherwise, the available material would put someone looking into a study close to par with the regulators. Says Tom Jefferson [in the second blog]:
The radical nature of the EMA’s policy lies in its offer of routine access to full clinical study reports [CSRs] with [hopefully] minimal redactions just a few days after drug approval [or rejection]. CSRs offer us the possibility of delving into and cross checking all aspects of trials: from protocol to informed consent forms, from intended analysis methods to those which were actually used, to templates for participant data collection. Some other aspects of the policy and its implementation are revolutionary, such as access to CSRs of drugs that did not make it to the European market. The thousands of pages provide a hitherto rarely tapped bonanza of detail and the possibility to address reporting bias — the biggest threat to contemporary, ethical research synthesis.
Tom addresses several implications of this "revolution" in the second blog which I’ll leave for you to read on your own except for one. What about the FDA?
Regulators will also be in a difficult position, with the Food and Drug Administration [FDA] left looking like a stranded whale wrapped in a sort of pre-New Deal commercial secrecy isolationism. The FDA does not release CSRs and the public can only access FDA reviews of the market authorisation application [which it calls NDAs or new drug applications]. These, like all reviews, do not tell you everything about all the trials in the programme, but represent the FDA’s take on the one or two pivotal trials that regulators looked at in depth with sponsors’ agreement. We tried to reconstruct a CSR narrative with sufficient detail to allow full appraisal from FDA medical officers reviews [MORs] in our neuraminidase inhibitors review. We failed completely because of a lack of sufficient information in the MOR and other documents available from Drugs@FDA. Use of FDA MORs instead of full CSRs risks swapping one incomplete source [journal articles] for another [FDA reviewers’ comments].
When I first started looking into clinical trials and discovered Drugs@FDA and the NDAs, I was pretty impressed. I could find out things that went beyond just looking at the journal articles. I submitted a few FOIA requests, and fairly quickly received a disc with what I was looking for. But those early explorations were of drugs that had been approved a long time ago. And Dr. Jefferson’s right, what you get depends on the reviewer. When I tried to look at more recent drugs, things were less smooth. For one of the last requests I submitted, I got a call from the FDA and the lady tried to talk me out of making the request in the first place. When I refused and asked her how long it would take, she said about two years [that was three years ago] and I never heard from them again. As he says, they’re going to look pretty foolish continuing to keep the PHARMA data secret when the EMA is being so transparent.
Along a similar note from the July 1, 2015 WSJ:
Doctors, Hospitals Got $6.49 Billion From Firms in’14
By Peter Loftus
And Joseph Walker
Who is your buddy, who is your pal, and what CFI’s?
“In its statement Tuesday, the AMA said “the vast majority of the data released today has not been independently validated by physicians, which makes it less usable for the patients its intended to benefit.”
So the AMA’s position is we need more time to ask every physician listed if they received the amount reported and then, and only then, can the information be used. This is the type of delaying effort one would expect from those that control the RUC as well as makes large sums of money from the continual updating of fees used in the computer billing industry.
Steve Lucas