BMJ Openby Peter Doshi and Tom Jefferson26 February 2013
Objective To explore the structure and content of a non-random sample of clinical study reports [CSRs] to guide clinicians and systematic reviewers.Search strategy We searched public sources and lodged Freedom of Information requests for previously confidential CSRs primarily written by the industry for regulators.Selection criteria CSRs reporting sufficient information for extraction [‘adequate’].Primary outcome measures Presence and length of essential elements of trial design and reporting and compression factor [ratio of page length for CSRs compared to its published counterpart in a scientific journal].Data extraction Data were extracted on standard forms and crosschecked for accuracy.Results We assembled a population of 78 CSRs [covering 90 randomised controlled trials; 144,610 pages total] dated 1991–2011 of 14 pharmaceuticals. Report synopses had a median length of 5 pages, efficacy evaluation 13.5 pages, safety evaluation 17 pages, attached tables 337 pages, trial protocol 62 pages, statistical analysis plan 15 pages and individual efficacy and safety listings had a median length of 447 and 109.5 pages, respectively. While 16 [21%] of CSRs contained completed case report forms, these were accessible to us in only one case [765 pages representing 16 individuals]. Compression factors ranged between 1 and 8805.Conclusions Clinical study reports represent a hitherto mostly hidden and untapped source of detailed and exhaustive data on each trial. They should be consulted by independent parties interested in a detailed record of a clinical trial, and should form the basic unit for evidence synthesis as their use is likely to minimise the problem of reporting bias. We cannot say whether our sample is representative and whether our conclusions are generalisable to an undefined and undefinable population of CSRs.
 One runs into the most interesting people in the oddest of ways, looking into the most unlikely of topics. Tom Jefferson and Peter Doshi are the Tamiflu guys, the ones that spent years running down an epidemiology story that needed to be told, and at least for me, began to unravel the torturous and kind of boring story of how Clinical Trials are cataloged and documented. Tom is a British Epidemiologist with the Cochrane Collaboration in Rome, Italy. Peter, his colleague, is on our side of the pond in Maryland and now an editor with the BMJ. But this blog isn’t about their interesting story with Tamiflu and other things. It’s about a topic they’ve tried to clarify for all of us – the Clinical Study Reports that become part of the record of Clinical Drug Trials – a should·be·simple topic that’s complex·and·confusing [perhaps deliberately so].
One runs into the most interesting people in the oddest of ways, looking into the most unlikely of topics. Tom Jefferson and Peter Doshi are the Tamiflu guys, the ones that spent years running down an epidemiology story that needed to be told, and at least for me, began to unravel the torturous and kind of boring story of how Clinical Trials are cataloged and documented. Tom is a British Epidemiologist with the Cochrane Collaboration in Rome, Italy. Peter, his colleague, is on our side of the pond in Maryland and now an editor with the BMJ. But this blog isn’t about their interesting story with Tamiflu and other things. It’s about a topic they’ve tried to clarify for all of us – the Clinical Study Reports that become part of the record of Clinical Drug Trials – a should·be·simple topic that’s complex·and·confusing [perhaps deliberately so].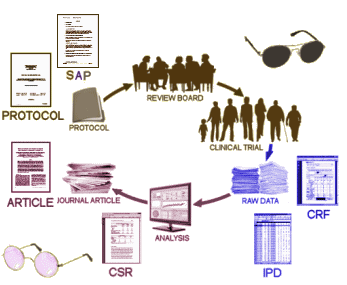
After a few years being preoccupied with evaluating the documents from a single Clinical Trial [Paxil Study 329] for a RIAT project [A Milestone in the Battle for Truth in Drug Safety, Restoring Study 329], I made this diagram trying to clarify things for myself. The upper part is clear – the a priori PROTOCOL and the SAP [Statistical Analysis Plan] – are evaluated by the Institutional Review Board [IRB] and, and if approved become the directives for the Clinical Trial. That part’s blinded [dark glasses]. Once the blind is broken, the Raw Data as Case Report Forms [CRFs] are assembled and organized. They’re transcribed into data tables known collectively as the IPD [Individual Participant Data]. Notice there aren’t any glasses there [yet] because these nuclear document have rarely ever been seen. Then someone uses the IPD to write an exhaustive CSR [Clinical Study Report]. Later all of this becomes the highly condensed published ARTICLE usually under the byline of academic authors. I put the glasses there because it’s seen and unblinded, but notice they’re "rose colored glasses." The important part is how to get untinted glasses in the IPD and CRF quadrant where they’re needed, without the "rose coloring" of conflict of interest:
Doshi and Jefferson explain some of why this eyes-on view of the Raw data is so hard to get hold of. The issue is that the meaning and requirements of CSR varies from company to company and agency to agency – and it’s not at all clear whether the CSR must contain the IPD [ergo, Raw Data] or not. In the study we looked at [Paxil Study 329], the IPD was listed in Appendices to the CSR, but in GSK’s initial release – the Appendices were nowhere to be found in the 2004 posting, only appearing 8 years later [2012] at the insistence of Peter Doshi working with the NY Attorney General. And apparently, there’s a lot of variability. So if this is a remote interest, by all means read their paper [maybe even if it isn’t, because this at the heart of the Data Transparency question].
Nice line: They’re not volunteering to be complicit with fraud. That’s all the more true when they do believe they are acting altruistically by signing up for a clinical trial.
Given the way these trials are done nowadays, I have my doubts whether anyone knows what happened to the patients overall. At most there might be people who each know about a small fraction of them — but they won’t be listed as authors, and they’ll probably have no control over how their results are described.
As for the subjects … I get a little nauseated reading about the drug cos’ concerns for them. As far as I can see, they are more and more being conned into believing that they’ve found a Clinic where they can get the care they have not been able to afford. The qualifying study on Abilify Maintena was “written” by Dr. John Kane in Glen Oaks NY, but the research was done at 49 different centers! Like this one in National City California:
http://www.synergyresearchcenters.com/depression-is-an-illness-not-a-weakness-2/
And this one in Flowood Mississippi:
precise-research.com
“Precise Research is one of the top depression clinics in Mississippi. Dr. Kwentus is one of the nation’s leading bipolar doctors. Precise also provides the best mental health treatment in the Jackson metro area and local region.” Hm. Begin your “Journey to mental health treatment’ here and you could end up in the ditch. Anyway, I bet none of the “authors” of that article have ever been to National City OR Flowood, and wouldn’t know Dr. Kwentus if they fell over him. Oh, but the drug company knows him!
Incredibly, this 329 trial only followed random people given this medication for 8 weeks at a time. If you’re only talking symptom suppression for several weeks, it can’t be too hard to find at least some outcome measures where people given medication do better than those on placebo for a short period.
These studies would mean so much more if they followed people for 1, 2, 3 years, and included more things that people care about – work functioning, social functioning, as well as control of distress (“symptoms”).
Maybe the companies sense that if they included long-term measures of social and occupational functioning, it would be laid even more bare how ineffective these pills are for “treating” life problems.
Edward, they aren’t setting out to “treat” life problems. They aim to treat persons with psychiatric diagnoses, which we know you consider invalid though you have not produced evidence or data to back that up. And what allows you to dismiss the subjects as “random people”? How do you justify that?
Yes, it would be good to have 3-year observation periods. Also, it would be good for everyone to win the lottery. There are practical constraints on clinical trials – time, money, subject fatigue leading to dropouts are the obvious ones. Where can we read about your 3-year trials of any treatment?
Measures of social and occupational functioning, as well as self-ratings by the patients, are in fact commonly obtained as secondary outcome measures. It’s always nice when these agree with the primary outcome measures that are based on clinician-rated severity. But in the Kabuki theater that the FDA has devolved into, it’s hard to blame the companies for focusing on primary outcome measures because that’s what the FDA rewards them for. It will take for two to tango if we want to correct the system.
Edward,
Actually, Study 329 did have a continuation phase [to 32 weeks]. And they did have three metrics aimed towards psycho-social function [autonomous function check list, sickness impact profile, and self perception inventory] all with no separation from placebo. The continuation phase, had a very high attrition rate besides the lack of separation making any meaningful analysis impossible. With a couple of exceptions, the a priori protocol for Study 329 had a more respectable design than many of the clinical trials of antidepressants [the protocol is in the beginning of Appendix A on-line].
Mickey,
Thanks for that information. I previously researched this study online and did not find any data for the 32 week mark, probably because of the elements you note here. Now I looked at the protocol link you gave and see that it was there. At least they tried to have a longer measure; that’s positive.
Bernard,
Thanks for your opinion. Here are a couple of articles that I read through recently on 3-5 year treatments:
http://archpsyc.jamanetwork.com/article.aspx?articleid=209673
http://www.madinamerica.com/wp-content/uploads/2014/12/open-dialogue-finland-outcomes.pdf
http://www.ncbi.nlm.nih.gov/pubmed/25900546 (this last one is the abstract only; but it’s a 2 year study of people labeled with various forms of psychoses)
To me, whether or not they are based on unreliable labels (which of course I think they are), this type of study is more interesting and encouraging than those following symptom control over a matter of weeks.
I hope you win the lottery 🙂
No cigar, Edward. The three reports you cited are not in the same ballpark as the trials Dr. Mickey was discussing.
The first compared two forms of psychotherapy for patients diagnosed with borderline personality disorder. The long duration is appropriate for that chronic condition but that is a different clinical context than registration trials of new drugs for a recent onset Axis I condition like major depression. Your second cite is an uncontrolled, naturalistic study of one particular treatment approach for psychosis using only a post hoc comparison against historical controls. It is in no way comparable to a new drug registration trial. Your third cite is an uncontrolled, naturalistic study of mediators and moderators of improvement in early psychosis. There was no comparison of treatments whatsoever.
In developing treatments, there is always a tradeoff between the ideal design and the pragmatic. The initial registration trials cannot be stretched out to 3 years of treatment, for the reasons I stated earlier. Candidate drugs that are duds can generally be screened out in trials that run for 2-3 months. A good example is the Corcept drug mifepristone that failed in Phase III trials for psychotic depression. Longer studies can be important and interesting at a later stage for agents that are approved. Pharmaceutical corporations will always try to game the system and to put lipstick on the pig, as the Pfizer reboxetine story and the GSK Paxil Study 329 story illustrate, but the solution there is open access to the data, not a 3-year registration trial length.
Now, what were you trying to say earlier by your mention of “random people”?
Bernard,
Luckily, studies don’t have to live up to the standards of an old Scrooge to mean something to the rest of us 🙂 These studies are different; that doesn’t mean they’re bad. They provide support for the idea that when provided with long-term family or individual psychotherapy, people with very serious emotional problems can do pretty well on functional outcome measures. Thank goodness some of these poor people have that option; in their position I’d take my chance on Open Dialogue or twice weekly psychotherapy, rather than hope for lasting results based on studies of pills lasting a few weeks.
As for my earlier comment, I was commenting on the lack of reliability behind the label of adolescent depression. If reliability ratings for adult depression are already relatively poor, one must wonder how reliable the ratings of distress in teenagers is going to be, with all the change occurring at that time of life. Depression is not one illness with a known etiology; rather, many totally different stresses can lead to different versions of the syndrome labelled depression. This makes it challenging to to have an objective reliable study about that label.
Of course, if I make that critique about the 329 study, I have to apply it also to the studies covering “schizophrenia” and “BPD”. Psychiatric research is much less precise than that of true sciences like physics or chemistry, because there are many uncontrolled variables that can confound any given study of human emotional / relational / developmental problems. These are quasi-experimental studies and so it’s better to look at large numbers of such studies to try to see broad trends, rather than looking at any individual study as if it meant much on its own. That’s my opinion.
Straw man alert. Who said the studies you cited are bad? But your citing of them was tangential.
When you keep changing the subject, a linear discussion is impossible. We were discussing standards of clinical trial design and oversight, then suddenly you veer off to state a tendentious preference for psychotherapy over medications, and to make adverse comparisons between clinical sciences and physics or chemistry – something that is not news and certainly not a reason to reject clinical science.
You recently offered some explicit advice to another commenter. Maybe you should follow your own advice.
“In developing treatments, there is always a tradeoff between the ideal design and the pragmatic. The initial registration trials cannot be stretched out to 3 years of treatment, for the reasons I stated earlier. Candidate drugs that are duds can generally be screened out in trials that run for 2-3 months.”
I find this so confusing. Given the long term nature of serious mental illness, how then does anyone ever know whether or not some people would have fared better without these medications? (I realize this may be a tangent to this post -but could it be discussed in another post )
Sally, it’s a process. It goes in stages. And keep in mind that the FDA isn’t there to advise on best practice in medicine. The FDA was mandated by Congress to regulate the claims of drug and device manufacturers as to basic efficacy and safety, that’s all. In registration trials, your question is addressed by having placebo treated control groups. Once a drug is registered based on such trials, then it is up to the professional groups to explore clinical issues like length of treatment, comparative efficacy against psychotherapy, longer term safety profile, possible preventive action, predictive biomarkers or gene markers, predictive psychological profiles, and so forth. At most, the FDA might direct the manufacturer to conduct Phase IV studies in particular populations such as the elderly or youth or pregnant women, all of whom are routinely excluded from standard Phase III registration trials. Because it is a process, we never know the absolute last word on a new medication until maybe 20 years after it is introduced. But we have to start somewhere with pragmatic indications of efficacy and safety.
Sally,
ditto. But you are correct that the “in use” part after a drug has been approved is in the hands of Medicine [the profession]. Those studies take a while and need to be financed. They have probably suffered in the modern era from the fall-off in public financing at the NIH/NIMH/VA etc. David Healy is addressing that in the area of harms with his Rxisk site – trying to model the kind of consumer feedback that would be useful for ongoing monitoring. A lot of that also comes when doctors follow their own patients, and the current HMO/Managed Care system doesn’t make that so very easy.
Bernard,
Can’t we just be friends? 🙂
Edward, is that the right way to frame the issue? When I make public comments I am not thinking about relationships but about the rigorousness of the discussion. And I am not interested in just repeating well-worn talking points but in moving the ball down the field. It’s that simple. So, when I challenge you or anyone else it’s because I am an equal opportunity critic, that’s all. I don’t view you as an enemy and, sure, we can be friends, and I will still call you out if I think I should. Fair enough?