So last week I had my best ever idea yet [they already don’t…], and Nick took pity on that post [thanks Nick] because it looked lonely with no comments. Undeterred, I’m having another shot at it. The gist of things is that I think we’re trying to intervene at the wrong level. Clinical Trials are now done by the Clinical Research Organizations and Clinical Research Centers. They have to be licensed to do human research. My plan is to make Clinical Trial Registration a requirement of their licensure. Pull their licenses if they do any Clinical Trial that’s unregistered. Part Two has to do with the data. Require that they send the same reports [anonymized] to the FDA as they send to the Clinical Trial Sponsor. Subject confidentiality should include the Sponsors too. Pull their licenses if they fail to file their reports with the FDA. What a great idea! So I decided to press on trying to document how big the non-registration, non-result-reporting problem actually has been, and I got bogged down until now. Then there it was on Jack Friday’s site [Pharmagossip]! I should pay him a finder’s fee for all the good stuff he digs up!
PLoS ONEby Vojtech Huser and James J. CiminoJuly 9, 2013
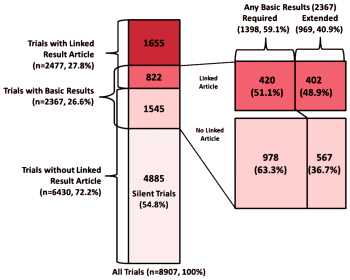
Objective: In an effort to understand how results of human clinical trials are made public, we analyze a large set of clinical trials registered at ClinicalTrials.gov, the world’s largest clinical trial registry.Materials and Methods: We considered two trial result artifacts: [1] existence of a trial result journal article that is formally linked to a registered trial or [2] the deposition of a trial’s basic summary results within the registry.Results: The study sample consisted of 8907 completed, interventional, phase 2-or-higher clinical trials that were completed in 2006-2009. The majority of trials [72.2%] had no structured trial-article link present. A total of 2367 trials [26.6%] deposited basic summary results within the registry. Of those, 969 trials [10.9%] were classified as trials with extended results and 1398 trials [15.7%] were classified as trials with only required basic results. The majority of the trials [54.8%] had no evidence of results, based on either linked result articles or basic summary results [silent trials], while a minimal number [9.2%/ report results through both registry deposition and publication.Discussion: Our study analyzes the body of linked knowledge around clinical trials [which we refer to as the “trialome”]. Our results show that most trials do not report results and, for those that do, there is minimal overlap in the types of reporting. We identify several mechanisms by which the linkages between trials and their published results can be increased.Conclusion: Our study shows that even when combining publications and registry results, and despite availability of several information channels, trial sponsors do not sufficiently meet the mandate to inform the public either via a linked result publication or basic results submission.hat tip to Pharmagossip
It’s a waste of time dealing with the pharmaceutical companies sponsoring clinical trials. They don’t want to make their raw data known for obvious reasons AKA "competitive advantage." If subject confidentiality matters, then they shouldn’t know the identity of the subjects either. So, remove them from the loop. File the raw data with the FDA when the blind is broken. All the FDA needs is some really big hard disks [which they already have]. The pharmaceutical companies don’t even bother to follow the rules already in place. What dreamer thinks they’ll follow any new rules?
Terrific idea, Mickey. Now let’s figure out how to get it past the far-right extremist politicians (who want no regulation) and all those other congresscritters (both left and right) who profit so handsomely from campaign contributions of BigPharma. It’s certainly a ‘do-able’ feat . . . I just don’t know if it’s doable in the current political environs.
What needs to be understood is that this is a process. You will not out gun pharma with one idea as they will lobby and work around it in a very short time.
This is a great concept, but you will need to look at international standards as pharma is already using studies performed in other countries for the basis of EU and American drug approvals. The phrase: We complied with all applicable laws will become all too common.
While we are all the same, we are all unique, and slight genetic variations have already caused serious medical side effects in some medications.
Getting data from drug and device companies will be a long hard slog that will never end as for every rule or law pharma will complain and lobby about so many rules and laws. Sadly, legal settlements in the deaths of children in places such as India and South America have shocked the countries involved into stricter study controls.
We can only hope that US and EU regulators realize this is a world wide issue and they need to look beyond their own borders to protect not only their citizens, but also innocents who are being misused for financial gain.
Steve Lucas
The FDA is not exactly the most computer-savvy organization. They still haven’t figured out how to mine their adverse event reports, and they’ve been collecting those for decades.