BMJ: Blogsby Stuart Buck30 Jan, 15… But for all the good that ethics review boards do, today they often block undeniably valuable research from going forward. The re-analysis of clinical trial data is a recent case where specious ethics objections are used to stymie good research into the effectiveness of drugs given to patients. The reason to re-analyze clinical trials is simple: far too many clinical trials are misreported or hidden from public view entirely. One recent study strikingly found that only 22 percent of trials obeyed the federal law requiring results to be reported publicly within a year of completion. To make matters worse, published clinical trials can still be highly misleading, because articles are often selectively written so as to highlight a desired finding. As a result, we are misled into overestimating the benefits, and underestimating the risks, of drugs.
The best way to ensure accurate information about all drug benefits and side effects is for all NIH-funded or FDA-regulated clinical trials to share patient level data with independent researchers. It is encouraging that the Institute of Medicine [IOM] recently issued a massive report recommending just that. In the IOM’s words, “Limited data sharing prevents maximum utilization of knowledge gained. …Greater data sharing could enhance public well-being by accelerating the drug discovery and development process, reducing redundant research, and facilitating scientific innovation.” Right on. Unfortunately, the IOM’s recommendations apply only to future clinical trials—2015 and beyond—not to what it calls “legacy” trials. To put this in context, as of the end of 2014, an all-time total of 1,496 drugs had been approved by the FDA, yet only 41 of those approvals occurred in 2014.
Thus, data sharing for future trials is good, but it is not enough. Virtually all drugs prescribed by doctors were studied in clinical trials prior to the IOM committee’s report in January 2015. Shouldn’t independent scholars be free to re-analyze those trials and see whether drugs are actually as effective and safe as promised? It seems like a no-brainer, but here’s where ethics committees rear their heads, reasoning that it would actually be unethical to re-analyze data unless the original patients sign another consent form. As an IOM committee member told The Wall Street Journal, “The primary challenge is the issue of informed consent. Patients who participated in trials in the past were most likely in trials that did not include [a provision for] sharing data publicly. So if we want to now share data, ethically, investigators should go back to get informed consent from the participants.”
Remember, no one is talking about re-doing a trial and exposing new patients to new drugs. The only question is whether the underlying data should be independently re-analyzed to see how well the drug actually worked. For anyone worried about privacy, there is no more reason to be worried about the independent analyst than about whoever did the original trial, and it is trivially easy to protect privacy in the same way that the original trialists did — sometimes even more so, now that we have software that permits someone to analyze data without ever downloading or seeing an individual patient’s records. Requiring patients to consent a second time is therefore unnecessary and irrelevant…
More importantly, what value will it be to the surviving patients to have to bother with yet another consent form? They already consented to be in a study, and they surely never meant for their data to be misused and misrepresented, as Ben Goldacre documents in his book Bad Pharma. No one has identified a single patient, in all of history, who has stated anything like the following:
When I signed up for that clinical trial, I was putting my health at risk and exposing my life to examination only for the benefit of one pharmaceutical company’s sales or one professor’s publication record. I never intended for any independent scholars to be able to double-check whether the drug actually works.If, as the IOM report suggests, some patients previously signed consent forms that actually ruled out future data-sharing, the data should still be subject to sharing. This is because those consent forms are what should be deemed unethical. After all, it is hard to imagine that any pharmaceutical company told patients:
You’re signing a form that guarantees that our paid analysts and ghostwriters will write about this data only if it is in our financial interest. Thanks to your signature, we will be able to convince “ethicists” that you didn’t want anyone to double-check our analysis, and we will thus be able to misrepresent the trial more effectively.Indeed, the very fact that anyone made patients sign such consent forms in the first place is a sign that the trial’s data could be especially in need of re-analysis. To be sure, it can still be difficult to re-analyze older clinical trials, due to software and formatting challenges. But when trial sponsors rely on the putative interests of patients in order to argue that data should be restricted, that is a direct obstruction to open science. Remember why patients made the sacrifice of participating in clinical trials in the first place: to generate accurate information about ways to cure disease. It is perverse to use those patients’ purported wishes as an excuse to allow misinformation to stand unchecked.
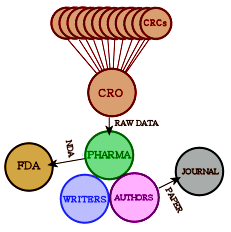 Obviously what we want is the raw data from the Clinical Trial – the same thing that the pharmaceutical company has to work with. In the good old days, only one agency was involved. Nowadays, the study sites [CRC – Clinical Research Centers] are coordinated and managed by a Clinical Research Organization who collects the data and passes it on to the PHARMA. The CRO/PHARMA analyze it and pass the summary to the writers. Once it’s drafted, the guest authors come into the picture and send it to a journal. If there is an FDA NDA submission, it comes from the PHARMA, prepared by some mixture of the CRO, writers, and PHARMA. What we see on the FDA web site is the FDA’s Reviewer report about the submission, but not the contents of the submission.
Obviously what we want is the raw data from the Clinical Trial – the same thing that the pharmaceutical company has to work with. In the good old days, only one agency was involved. Nowadays, the study sites [CRC – Clinical Research Centers] are coordinated and managed by a Clinical Research Organization who collects the data and passes it on to the PHARMA. The CRO/PHARMA analyze it and pass the summary to the writers. Once it’s drafted, the guest authors come into the picture and send it to a journal. If there is an FDA NDA submission, it comes from the PHARMA, prepared by some mixture of the CRO, writers, and PHARMA. What we see on the FDA web site is the FDA’s Reviewer report about the submission, but not the contents of the submission. 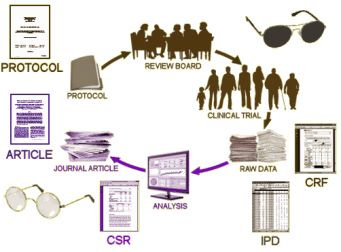
I don’t actually know what the NDA [New Drug Application] contains – summary data? the individual participant data [IPD]? clinical report forms [CRF]? is it the clinical study report [CSR]? But for the moment, the question is how does whatever consent the subject gives pass to the FDA to enforce? Where does the authority of the FDA to withhold the data they analyzed come from? We all know that the claim of privacy for the subjects is bogus, but the actual process of the system, the content of the consents, the authority of the FDA, and who has what is murky – at least to me. We’re mounting arguments, but they’re blowing in the wind because we [at least I] don’t know who we’re arguing with about what. I have the feeling the people in the Institute of Medicine are in the same boat. It seems to me that the first order of business is to get hold of those consent forms and see what they really say. And what about that «NOTE TO SELF»? Did the consent have a provision to give the data to the FDA? How is making the data available for research purposes to the PHARMA Sponsors or the FDA different from making the data available for research purposes to independent researchers? Is the consent to the CRC? the CRO? the PHARMA Sponsor? any researcher? Many questions…
The consent form issue is a straw man argument designed to delay, delay, delay. As the information becomes available and patients and regulators see patterns pharma is well aware all of their trials will become questionable.
The end game for pharma is “What does it matter” as drugs go off paten, complaints grow old, patients die, and this becomes yesterday’s news allowing the system to continue on as it has in the past.
Steve Lucas
We don’t need no stinkin’ ethics.
As someone who has seen raw data coming directly out of clinical trials, I can say the individual patients often are identified by opaque code numbers — the code numbers link to patient information in another document or database, breaking the blind.
If somehow the code numbers are meaningful, identifying birthdates, for example, they can be re-coded or even randomized by adding another database table.
If identifying patients by code numbers has not been done in a clinical trial, it would be trivially easy to do so by interposing another database table of code numbers between patient information and the raw data tabulations.
So that part of the argument is invalid.
I would think that where a consent agreement includes a provision for release of anonymized data, this would be sufficient for data sharing.
In other situations, an ethics panel would have to determine if patient privacy is breached by release of anonymized data. Personally, I can not see how a patient is harmed in this way.
A universe of 1,496 drugs with x studies per drug is not infinite. Such analysis of consent agreements is not impossible.