A Phase 3 Randomized Clinical Trial [RCT] of a medication isn’t really research. That’s already been done earlier. The RCT is product testing with two important elements – efficacy [does it work?] and safety [does it cause harm?] – and in either case, what is the strength of the effect? Other things might become apparent along the way. Who knows? Maybe some day the secret of the universe might be serendipitously found in an RCT, but that’s still an incidental, exploratory finding – not why the trial is being done.
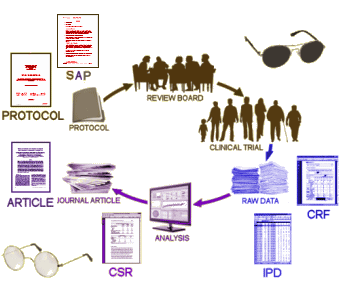
Everything about a given trial is specified in two documents [often combined] – the Protocol and the Statistical Analysis Plan – prepared in advance [a priori] and submitted to an Institutional Review Board [IRB]. After the Clinical Trial is approved and the blinded study is underway, no changes can be made except under unusual circumstances, and then only by an official amendment to the Protocol approved by the IRB.
Why so rigid? It’s obvious. There’s never any guarantee that the sanctity of the blind hasn’t been breached, informing any after·the·fact changes. Anyone who has any experience with statistical analysis knows that with foreknowledge, one can have a field day with statistical testing. Shuffle some outcomes, select the right tests, ignore the negatives, etc. And that’s exactly what has happened – over and over. Weak signals without clinical significance have been turned into lucrative blockbusters. While it’s hard to prove, it can easily be prevented.
Our primary defense against such perversions of scientific reporting is fidelity to the registered IND protocol and plan of statistical analysis. The solution is not hard to see: We need independent analyses of clinical trials because we cannot trust the corporate analyses. In effect, we need something like the Underwriters Laboratory to verify the statistical analyses of clinical trials. Nobody takes the manufacturing corporation’s word for it concerning the safety and performance of X-ray machines or cardiac defibrillators. Why treat the statistical analysis of drug trials any differently? It’s highly technical work. Who should assume that responsibility? Why not the FDA? After all, they alone see all the data. My specific proposal is for Congress to mandate that the FDA analyze all clinical trials data strictly according to the registered protocols and analysis plans. That requirement should apply to new drugs or to approved drugs being tested for new indications. It should apply also to publications reporting new trials of approved drugs. Corporations and investigators should be prohibited from publishing their own in-house statistical analyses unless verified by FDA oversight.
As originally conceived, the ECDEU program consisted primarily of grant- supported clinical investigators working in tine common area of psychotropic drug evaluation [both new and established compounds]. One of the problems they encountered, and task they accomplished, was the development of a uniform battery of clinical assessment instruments known as the ECDEU Standard Reporting System, first introduced for utilization in 1967. The rationale behind this effort was twofold. First, it was felt that such a system would enhance both the quality of early clinical drug research and allow greater generalizability of results across studies and investigating units. Second, data collected on common forms could be stored in a data bank for future study and research. Since the implementation of this Standard Reporting System and the Biometric Laboratory Information Processing System [BLIPS], the ECDEU program has evolved into more than an extramural grant support program for psychotropic drug research teams. In collaboration with The George Washington University Biometric Laboratory, the ECDEU Standard Reporting System has been made available to any investigator interested in conducting clinical trials, whether federally grant supported or not. To utilize these services, the investigator is requested to:
1. Submit a Research Plan Report and agree to send the study data to the Biometric Laboratory.2. Collect sufficient information about the subjects in his study so that the data can be entered into the ECDEU data bank. This means, essentially, that a core of data must be collected for each patient…
In return, he receives a sufficient number of assessment scales to conduct his research. Once the trial is completed, the forms are returned to the Biometric Laboratory for processing and data analyses, the results of which are sent to the investigator in the form of a standard data package. The rating scales and data processing services are provided at no charge – our sole "remuneration" being the opportunity to add the investigator’s data to the data bank. It should be stressed that an investigator’s data and/or results are never published or disseminated to others without his permission.
Sorry, the comment form is closed at this time.