Posted on Saturday 5 July 2014

ChemistryWorldby Andy Extance3 July 2014Tempers are being tested as the pharmaceutical industry’s journey towards transparency on clinical trial data enters a critical phase. Even as drug companies announce more voluntary access schemes, campaign group AllTrials has accused the European Medical Agency [EMA] of a ‘backroom deal with pharma’ to weaken earlier commitments. The disputes have arisen following EMA consultations over its clinical trial data policy, which it hopes to finalise in mid-July, and bring into force in October. The pioneering plan would be the first of its kind to proactively publish clinical trial reports, giving people access for non-commercial use without needing to request it from companies. After receiving over 1100 comments covering a broad spectrum of opinions, the EMA conducted follow-up meetings in May to discuss the resulting proposals. Following the meetings researchers, AllTrials, and even the EU Ombudsman reacted critically.
The chief concern was a ban on saving, downloading or printing clinical study reports [CSRs], making these huge documents available only on-screen. The Institute for Quality and Efficiency in Health Care [IQWiG], Germany’s national commission for assessing medical procedures, was especially outraged. It took to Twitter with a long series of photos underlining how hard this would make its job. The same groups also warned that the new ‘terms of use’ contract could let trial sponsors sue researchers, creating a legal chill that would deter scrutiny.
The EMA’s new redaction policy on what information could be hidden drew fire too. It arose because of comments in the initial consultation that patient privacy, trust in the system, and commercial confidentiality could be put at risk. Richard Bergstrom, director general of the European Federation of Pharmaceutical Industries and Associations [EFPIA], expressed worry over ‘putting transparency – at whatever cost – ahead of public health interests’.
The proposed EMA policy allows drugmakers to propose redactions for text they feel is commercially or otherwise sensitive, although the regulator will retain the final say on what is hidden. Campaigners say this measure and vague policy wording will mean too much is concealed. EMA spokesman Martin Harvey-Allchurch stresses that ‘third parties’ can petition or sue if they think that’s happening, and that the extent of redaction will always be visible.
Working too closely?Yet the new policy still seemed to many a significant departure from the EMA’s previous hard line on data disclosure. Its 2010 ‘access to documents’ policy considers that most CSRs are not commercially confidential information, and promised to disclose CSRs for every drug it had reviewed. The confidentiality point has drawn fire from some drugmakers, with AbbVie and InterMune disputing it in court. But in April, AbbVie dropped its case, the day after the EU passed regulations that will make clinical trial registration and data sharing a legal requirement. Then, at the end of May, InterMune dropped some of its cases against the EMA.
AllTrials subsequently suggested that the price for dropping the cases had been a more industry-favourable transparency policy, something Harvey-Allchurch ‘absolutely refutes’. ‘AbbVie realised that we’re going to stick with our definition of commercial confidentiality – they knew they were not going to win that one,’ he says. ‘They went away, and came back with proposals for what they wanted redacted. In the end, what they proposed was in line with our redaction principles. The main InterMune case is still running.’
Following the pressure over its proposals on 12 June, the EMA’s management board agreed an amended policy. That allows downloading, saving and printing trial data for academic and non-commercial research purposes, although the other controversial areas remain unchanged. Nevertheless Harvey-Allchurch plays down the outcry surrounding the new policy. ‘The whole purpose of these targeted consultation exercises was to listen,’ he says. ‘We heard the feedback, we listened to it, and proposed an alternative to the board.’ But with Harvey-Allchurch underlining that this policy initiative is a bridge to the new EU regulations that will come in some time after May 2016, the final policy wording will be highly significant.
Industry deliversMeanwhile, pharma companies continue to progress their own schemes to make data available, with Bristol-Myers-Squibb [BMS] becoming the latest to open up its trial results. The Duke Clinical Research Institute at Duke University in Durham, North Carolina will act as the gatekeeper for BMS’ data. It will review study proposals and check final manuscripts ‘for scientific integrity and consistency with the original proposed work’ like Yale is doing for Johnson & Johnson. Boehringer-Ingelheim has also said that it will publish documents for all approved products going back to 1998. That move goes beyond the ‘Principles for responsible clinical trial data sharing’ introduced by the EFPIA and the Pharmaceutical Research and Manufacturers of America [PhRMA] last July.
IQWiG spokesperson Susanne Breuer highlights that a fragmented landscape of company data repositories would be less desirable than the single repository the EMA hosts. ‘The control of data completeness would be much more complicated, and analysing data it would take a lot longer,’ she says. ‘We would appreciate centralised access for scientists.’ Parts of the industry are making progress here, with Eli Lilly and Bayer Healthcare agreeing to make trial data available via Clinicalstudydatarequest.com. They bring the number of companies using the portal, created by GlaxoSmithKline last year, to eight.
 I first became aware that PHARMA was giving up on CNS drug development three years ago when Dr. Stahl let loose with a rant of cosmic proportions [
I first became aware that PHARMA was giving up on CNS drug development three years ago when Dr. Stahl let loose with a rant of cosmic proportions [
 Were Dr. Insel able to transcend his fixation on Clinical Neuroscience and the as-yet-unfound-novel-breakthroughs-around-the-corner he’s chasing, and could sit quietly under the Bodhi tree beside the river, what faint music might he hear building in the stillness as he mused on his ko
Were Dr. Insel able to transcend his fixation on Clinical Neuroscience and the as-yet-unfound-novel-breakthroughs-around-the-corner he’s chasing, and could sit quietly under the Bodhi tree beside the river, what faint music might he hear building in the stillness as he mused on his ko set of lens than the the rose colored glasses that came with the passion of new discovery. But what’s supposed to happen rarely does happen. The proponents try to keep the dream alive, and in the process add a dark patina that increases and prolongs the depth of the period of disillusionment.
set of lens than the the rose colored glasses that came with the passion of new discovery. But what’s supposed to happen rarely does happen. The proponents try to keep the dream alive, and in the process add a dark patina that increases and prolongs the depth of the period of disillusionment.
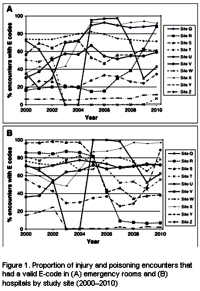 While I’m still grumbling about the Lu et al papers [3], my main agenda in this post is something else – the use of these large databases in research papers. The story begins with a problem – the commercial databases don’t have usable E-Codes, the ICD-9CM codes that directly address suicide attempts. In their Letter to the Editor [3], Lu et al prove that the database they are using doesn’t have these E-Codes with any consistency across sites. So they bring up Patrick et al [2] who did a study trying to deal with the missing E-Codes by locating other parameters that could be used as a proxy for suicide attempts. They tried a number of combinations and came up with an algorithm involving psychiatric diagnosis and injury to the lower arm or asphyxiation or overdose with psychotropic medications using two government databases that had required E-Codes as their gold standard.
While I’m still grumbling about the Lu et al papers [3], my main agenda in this post is something else – the use of these large databases in research papers. The story begins with a problem – the commercial databases don’t have usable E-Codes, the ICD-9CM codes that directly address suicide attempts. In their Letter to the Editor [3], Lu et al prove that the database they are using doesn’t have these E-Codes with any consistency across sites. So they bring up Patrick et al [2] who did a study trying to deal with the missing E-Codes by locating other parameters that could be used as a proxy for suicide attempts. They tried a number of combinations and came up with an algorithm involving psychiatric diagnosis and injury to the lower arm or asphyxiation or overdose with psychotropic medications using two government databases that had required E-Codes as their gold standard. 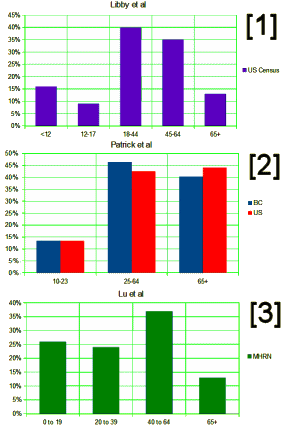
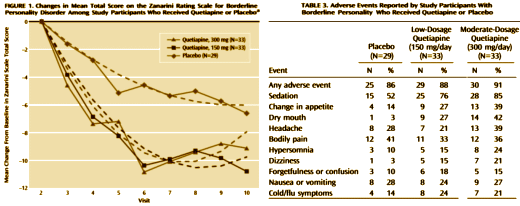

 And the Seroquel XR® study is no different, having no significant effect on core symptoms [speaking of significance, the higher dose Seroquel XR® was not statistically significant]. This study is a classic "experimercial" aiming towards fostering the off-label use of Seroquel XR® in this condition. This whole enterprise of doing Clinical Trials in Borderline Personality Disorder is not science based, more a shotgun approach to see what sticks. Here’s another one from four years ago, funded by Eli Lilly aiming to study Zyprexa in this group of patients, also by Dr. Schulz. Similar results:
And the Seroquel XR® study is no different, having no significant effect on core symptoms [speaking of significance, the higher dose Seroquel XR® was not statistically significant]. This study is a classic "experimercial" aiming towards fostering the off-label use of Seroquel XR® in this condition. This whole enterprise of doing Clinical Trials in Borderline Personality Disorder is not science based, more a shotgun approach to see what sticks. Here’s another one from four years ago, funded by Eli Lilly aiming to study Zyprexa in this group of patients, also by Dr. Schulz. Similar results: 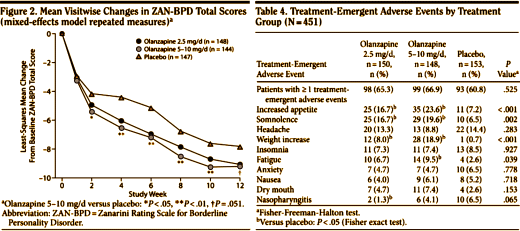
 -n
-n k
k r
r z
z
 It’s time for our journals to realize that they’re not roadside billboards and put an end to publishing articles whose primary purpose is advertising expensive drugs, off-label, particularly the major journals like the American Journal of Psychiatry. That’s just not what they’re for. That’s never been what they are for…
It’s time for our journals to realize that they’re not roadside billboards and put an end to publishing articles whose primary purpose is advertising expensive drugs, off-label, particularly the major journals like the American Journal of Psychiatry. That’s just not what they’re for. That’s never been what they are for…