This email response to a researcher who was requesting funding from Zeneca several months after the F.D.A.’s approved Seroquel might seem odd or even Machiavellian to a Basic Scientist, a Practicing Clinician, or a patient-to-be, but if your business is selling the product, it makes perfect sense:Zeneca had poured years and a lot of money into getting their drug approved. Now it was time to focus on reaping the benefits of their hard work…

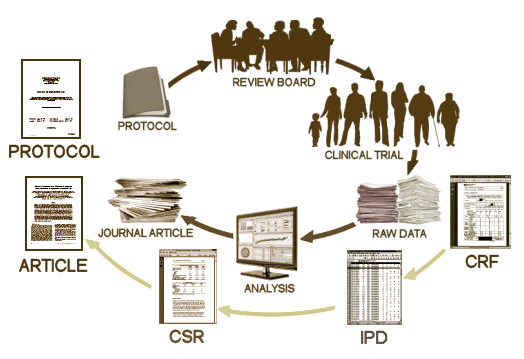

 The PROTOCOL is a formal document that lays out in detail how the study is to be conducted. It has multiple functions, but for our purposes, it’s important because there are a number of ways that a study can be skewed in favor of a drug – like picking a dose for the comparator drug that is either to low to be effective or to high and likely to cause a number of side effects and discontinuations. It’s important because it lays out the primary and secondary variables and how they will be analyzed. All of this is a priori, before the study is done. One reason is that given enough creativity, one can frequently find significance after the fact by running tests on any and everything. By making a priori declarations of both target and technique, the PROTOCOL assures us that we’re not being taken down some after-the-fact garden path. The PROTOCOL is an essential element for Data Transparency.
The PROTOCOL is a formal document that lays out in detail how the study is to be conducted. It has multiple functions, but for our purposes, it’s important because there are a number of ways that a study can be skewed in favor of a drug – like picking a dose for the comparator drug that is either to low to be effective or to high and likely to cause a number of side effects and discontinuations. It’s important because it lays out the primary and secondary variables and how they will be analyzed. All of this is a priori, before the study is done. One reason is that given enough creativity, one can frequently find significance after the fact by running tests on any and everything. By making a priori declarations of both target and technique, the PROTOCOL assures us that we’re not being taken down some after-the-fact garden path. The PROTOCOL is an essential element for Data Transparency.  The CRF‘s [CASE REPORT FORMS] are the primary raw data source, and may be hundreds of pages long for each research subject. They are the various forms filled out by the study coordinators and have intake information, the actual forms of the subjects tests, the trial staffs’ recording of adverse events, the medication records etc. All of this information is recorded before the blind is broken. Things like adverse reactions are recorded in plain english rather than coded by some system. This is the data in its rawest available form [see below]. This information would be required for any serious vetting of adverse events for reasons stated below.
The CRF‘s [CASE REPORT FORMS] are the primary raw data source, and may be hundreds of pages long for each research subject. They are the various forms filled out by the study coordinators and have intake information, the actual forms of the subjects tests, the trial staffs’ recording of adverse events, the medication records etc. All of this information is recorded before the blind is broken. Things like adverse reactions are recorded in plain english rather than coded by some system. This is the data in its rawest available form [see below]. This information would be required for any serious vetting of adverse events for reasons stated below.
 T
T he IPD [INDIVIDUAL PARTICIPANT DATA] is the compilation of the data from the CRFs in tabular form, spreadsheets in one format or another, data transposed from the CRFs and ready for analysis [separated by treatment eg after the blind has been broken]. For an analysis of the efficacy data, this is all that would be required. The adverse event data is also usually in tabular form in either an abbreviated or encoded in some standardized way. If there are no questions about adverse events, the IPD tables would be fine for a reanalysis. But if adverse event data is in question, the actual CRFs are required reading to avoid lost-in-translation errors, as tedious as that might be.
he IPD [INDIVIDUAL PARTICIPANT DATA] is the compilation of the data from the CRFs in tabular form, spreadsheets in one format or another, data transposed from the CRFs and ready for analysis [separated by treatment eg after the blind has been broken]. For an analysis of the efficacy data, this is all that would be required. The adverse event data is also usually in tabular form in either an abbreviated or encoded in some standardized way. If there are no questions about adverse events, the IPD tables would be fine for a reanalysis. But if adverse event data is in question, the actual CRFs are required reading to avoid lost-in-translation errors, as tedious as that might be.
 The CSR [CLINICAL STUDY REPORT] is a long narrative document that tells the story from start to finish including the results. Sometimes it contains the actual data [IPD] as an Appendix and sometimes it has only summary tables. It’sa version of the published paper in the long form – typicallyseveral hundred pages. Depending on what questions are being asked and how thoroughly they include the IPDs, it might be adequate for checking a study – or it may not. It’s the report that industry would like to be Data Transparency, but it’s usually not close enough to the Raw data to suffice.
The CSR [CLINICAL STUDY REPORT] is a long narrative document that tells the story from start to finish including the results. Sometimes it contains the actual data [IPD] as an Appendix and sometimes it has only summary tables. It’sa version of the published paper in the long form – typicallyseveral hundred pages. Depending on what questions are being asked and how thoroughly they include the IPDs, it might be adequate for checking a study – or it may not. It’s the report that industry would like to be Data Transparency, but it’s usually not close enough to the Raw data to suffice.

 The published ARTICLE is what we’re used to seeing. There was a time in my lifetime that we thought that what we read in our journals was the real deal, but that time has passed. The many ways that data has been manipulated, misrepresented, jury-rigged, seen through rose-colored glasses has become a source of daily amazement to me in my retirement years. As I’ve often said, never in my wildest dreams. There are so many examples that industry really doesn’t have much of a legitimate argument for their claim of proprietary ownership of Clinical Trial data. Their record of gross abuse of that privilege is self-indicting.
The published ARTICLE is what we’re used to seeing. There was a time in my lifetime that we thought that what we read in our journals was the real deal, but that time has passed. The many ways that data has been manipulated, misrepresented, jury-rigged, seen through rose-colored glasses has become a source of daily amazement to me in my retirement years. As I’ve often said, never in my wildest dreams. There are so many examples that industry really doesn’t have much of a legitimate argument for their claim of proprietary ownership of Clinical Trial data. Their record of gross abuse of that privilege is self-indicting. I know this is all repetitive and tedious. If you already know it, it’s boring. If you don’t know it, it’s still boring. But the European Medicines Agency’s decision and Canada’s Vanessa’s Law [see doing the right thing…] are the two concrete markers for the cause of Data Transparency. There are lots of questions about Clinical Trials and their ultimate value, but right now the point is anterior to those considerations – basic honesty in scientific reporting. The tools of statistics, analysis, and presentation have been grossly perverted too frequently, for too long.
Just having the raw data available is of no value if someone doesn’t analyze published studies that are in question. Having done a some of that in the years between my naivete four years ago and the present, I can assure you that it’s not for the faint of heart. People who take on the task of vetting someone else’s data need to be well versed in modern analytic techniques or at least willing to take the time to get up to speed. If reading this post is tedious and boring, it’s a piece of cake compared to doing a careful analysis of a Clinical Trial. But the evidence of the last quarter century dictates that there is really no other choice if physicians are to be accurately informed about the medications they prescribe and patients are to know what they’re taking. So when the EMA policy comes out next month, read it carefully to see what is actually going to be available. It matters, and the devil is in the details…
To wit:
Pharmalotby Ed SilvermanAug 28, 2014Amid ongoing debate over the extent to which clinical trial data should be divulged, a new survey finds that an overwhelming majority of members of the Royal College of Physicians in the U.K. believe that such information should be disclosed and accessible. To wit, 95 percent say all trials should be registered; 89% says increased publication of results, including those that are negative, will lead to better medicines and patient healthcare; 81% agree that a “moral duty” exists for drug makers to make completed data available to trial participants, the public and the scientific community; and 87% says increased scrutiny of data will lead to better science and research.
At the same, time 10% believe increased publication and dissemination of clinical trial results will harm commercial interests of drug makers and only 18% say that increased access to trial data will harm commercial interests. Just 5% believe companies should not be required to release clinical trial data into the public, and only 27% say publication of completed data should be linked to market authorization.
“The world has changed,” writes Keith Bragman, president of the Royal College’s Faculty of Pharmaceutical Medicine, a standards-setting body at the Royal College, in remarks accompanying the survey results. “Society now demands greater transparency in clinical trials.”
Data disclosure, you may recall, has been a contentious topic following scandals over safety of effectiveness data that was not publicly shared. The survey, which queried 430 of the faculty’s 1,500 members, comes as regulators, academic researchers and drug makers dicker over policies for releasing trial data. The issue has been particularly fraught in Europe, where the European Medicines Agency has repeatedly delayed the release of its new policy amid criticism the agency has backtracked on a previous commitment to new-found openness. Last month, the regulator indicated formal adoption is scheduled to take place at an October board meeting…
seeing abbvie spam CNN w/ a variety of Humira ads, each of which had a different application/spin on the perception of the symptoms being treated, i hope the EMA steps up and delivers.