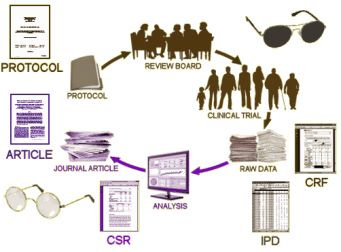
 We all know, that’s not what happens. The results are turned over to the corporate sponsor who analyze the results and use medical writers to produce the article. In many cases, the physicians and other scientists on the Author By-Line are primarily a ticket into a peer-reviewed journal, and have little if any input into the actual production of the article. And since the reader of the article can’t see any of the process that goes into the creation of what they’re reading, the data can be subtly manipulated to accentuate the positive and eliminate the negative. It not only can happen, it has happened in epidemic proportions – yielding enormous profits and more importantly has resulted in doctors prescribing many ineffective or sometimes harmful medications. The time for simply decrying this state of affairs has long passed. It’s time to put a stop to it, but that’s proving to be a very thorny task.
We all know, that’s not what happens. The results are turned over to the corporate sponsor who analyze the results and use medical writers to produce the article. In many cases, the physicians and other scientists on the Author By-Line are primarily a ticket into a peer-reviewed journal, and have little if any input into the actual production of the article. And since the reader of the article can’t see any of the process that goes into the creation of what they’re reading, the data can be subtly manipulated to accentuate the positive and eliminate the negative. It not only can happen, it has happened in epidemic proportions – yielding enormous profits and more importantly has resulted in doctors prescribing many ineffective or sometimes harmful medications. The time for simply decrying this state of affairs has long passed. It’s time to put a stop to it, but that’s proving to be a very thorny task.
Every solution to this problem starts with Data Transparency. Unless science at large has access to every part of that top figure, the problem will persist. And the industry that stands to gain has mounted a sustained resistance at every point along the road. Their public arguments are commercially confident information and patient confidentiality, but that’s simply a smoke-screen for their real problem. Without the cover of darkness, many of the medications released in recent decades wouldn’t have made it to first base. There’s nothing scientific about the idea that new drugs are better, or that new drug discovery can proceed at a predictable pace. "A good man drug is hard to find. You often always get the other kind." – wouldn’t be a bad theme song for this industry.
NIH Director’s Blogby Drs. Kathy L. Hudson and Francis S. CollinsNovember 19, 2014When people enroll in clinical trials to test new drugs, devices, or other interventions, they’re often informed that such research may not benefit them directly. But they’re also told what’s learned in those clinical trials may help others, both now and in the future. To honor these participants’ selfless commitment to advancing biomedical science, researchers have an ethical obligation to share the results of clinical trials in a swift and transparent manner.
But that’s not the only reason why sharing data from clinical trials is so important. Prompt dissemination of clinical trial results is essential for guiding future research. Furthermore, resources can be wasted and people may even stand to be harmed if the results of clinical trials are not fully disclosed in a timely manner. Without access to complete information about previous clinical trials — including data that are negative or inconclusive, researchers may launch similar studies that put participants at needless risk or expose them to ineffective interventions. And, if conclusions are distorted by failure to report results, incomplete knowledge can eventually make its way into clinical guidelines and, thereby, affect the care of a great many patients. Unfortunately, the timely public reporting of results has not been consistent across the clinical trials enterprise. For example, a recent analysis of 400 U.S. clinical trials found that even four years after the trials had been completed, nearly 30% had failed to share results by publishing in a scientific journal or reporting in ClinicalTrials.gov, a public database maintained by NIH.
Today, the Department of Health and Human Services [HHS] proposed a rule to require public sharing of key results — the summary data — from certain clinical trials of drugs and devices regulated by the Food and Drug Administration [FDA]. While such a mandate has been in place for several years, the proposed rule aims to clarify the requirements. Summary data would include: baseline characteristics of participants, primary and secondary outcome results, and information about adverse events. With a few exceptions, such results must be submitted to ClinicalTrials.gov within one year of the time when the trial completes collection of primary outcome data. But we at NIH are proposing to go one step further to address this important issue. Today, NIH released a draft policy for public comment to apply these data reporting requirements to all interventional clinical trials that it funds. We are committed to working with NIH-supported researchers and institutions to ensure the new responsibilities in this proposed policy are understood and any unanticipated obstacles are removed.
What all this really comes down to is trust. Clinical trial participants trust that the data they provide will be used by biomedical science to advance the health of many. Researchers seek to add to the body of biomedical knowledge and can respect this commitment by promptly sharing clinical trial data. There is also the important matter of public trust. American taxpayers trust NIH to be a good steward of their investment — and one way we can do that is to ensure that results of all our clinical trials are shared with the worldwide scientific community in an open and efficient manner. Data sharing is essential to turn even more scientific discoveries into better health at an even faster pace!
 This history has been written by many well-meaning congressmen, reformers, and agencies, but they haven’t stayed at it. Industry is always at it. So the crises pass and drug companies just keep on doing what they’ve been doing as soon as the dust settles. It’s time for a new clamor – enforcement and ongoing monitoring. Without those two things, it’s all just more empty promises…
This history has been written by many well-meaning congressmen, reformers, and agencies, but they haven’t stayed at it. Industry is always at it. So the crises pass and drug companies just keep on doing what they’ve been doing as soon as the dust settles. It’s time for a new clamor – enforcement and ongoing monitoring. Without those two things, it’s all just more empty promises…
| What is the Results Database? |
|
The ClinicalTrials.gov results database was launched in September 2008 to implement Section 801 of the Food and Drug Administration Amendments Act of 2007 [FDAAA 801] [PDF], which requires the submission of "basic results" for certain clinical trials, generally not later than 1 year after their Completion Date [see Primary Completion Date on ClinicalTrials.gov]. The submission of adverse event information was optional when the results database was released but became required in September 2009. Results information for registered and completed studies is submitted by the study sponsor or principal investigator in a standard, tabular format without discussions or conclusions. The information is considered summary information and does not include patient-level data. The results information that is submitted includes the following:
ClinicalTrials.gov staff review results submissions to ensure that they are clear and informative prior to posting to the Web site. However, ClinicalTrials.gov cannot ensure scientific accuracy. Data providers are responsible for ensuring that submitted information is accurate and complete.
|
| Commenting on the NPRM and proposed NIH Policy |
|
The public may comment on any aspect of the NPRM or proposed NIH Policy. Written comments on the NPRM should be submitted to docket number NIH-2011-0003 at www.regulations.gov [not there as of today?] Commenters are asked to indicate the specific section of the NPRM to which each comment refers. Written comments on the proposed NIH Policy should be submitted electronically to the Office of Clinical Research and Bioethics Policy, Office of Science Policy, NIH, via email at:
mail: at 6705 Rockledge Drive, Suite 750, Bethesda, MD 20892, or by fax: at 301-496-9839. The agency will consider all comments in preparing the final rule and final NIH Policy.
|
UPDATE: This is the working link for comments:
Here is language from 2007 concerning penalties for failing to submit clinical trials information and for submitting information that is false or misleading. It can be found on page 98 of this document.
PROHIBITED ACTS.—Section 301 of the Federal Food,
Drug, and Cosmetic Act (21 U.S.C. 331) is amended by adding
at the end the following:
‘‘(jj)(1) The failure to submit the certification required by section
402(j)(5)(B) of the Public Health Service Act, or knowingly submitting
a false certification under such section.
‘‘(2) The failure to submit clinical trial information required
under subsection (j) of section 402 of the Public Health Service
Act.
‘‘(3) The submission of clinical trial information under subsection
(j) of section 402 of the Public Health Service Act that
is false or misleading in any particular under paragraph (5)(D)
of such subsection (j).’’.
(2) CIVIL MONEY PENALTIES.—Subsection (f) of section 303
of the Federal Food, Drug, and Cosmetic Act (21 U.S.C. 333),
as redesignated by section 226, is amended—
(A) by redesignating paragraphs (3), (4), and (5) as
paragraphs (5), (6), and (7), respectively;
(B) by inserting after paragraph (2) the following:
‘‘(3)(A) Any person who violates section 301(jj) shall be subject
to a civil monetary penalty of not more than $10,000 for all violations
adjudicated in a single proceeding.
‘‘(B) If a violation of section 301(jj) is not corrected within
the 30-day period following notification under section 402(j)(5)(C)(ii),
the person shall, in addition to any penalty under subparagraph
(A), be subject to a civil monetary penalty of not more than $10,000
for each day of the violation after such period until the violation
is corrected.’’
So far, I have not been able to find language concerning tougher enforcement of these penalties in the new announcement. I would love to be corrected on this point.