I wish that what I write about could always be interesting, but I just can’t bring it off. Writing is my way of gathering my thoughts, and sometimes that’s pretty boring, even to me. My daughter who named this blog has apologized for her sarcasm, but to be honest, I’m very appreciative. The injunction to be interesting is just too daunting. So here’s some more details about that last post – what that EMA email clarified about what regulatory documents they are actually going to make available. It’s a two phase process, and we’re in phase 1. All we have right now from the clinical drug trials are the registration documents from a registry like clinicaltrials.gov and a published paper [if one is even published]. The inadequacy of that level of access is legend.
What the European Medicines Agency is planning to make available:
I’ve used some version of this diagram to relate the documents to the actual process of the trial itself.
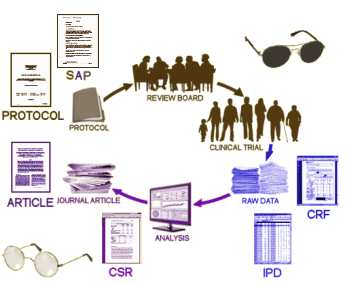
The PROTOCOL and the SAP [STATISTICAL ANALYSIS PLAN] are the two major documents that are a priori [written before the trial begins] and shouldn’t be changed without good reason along the way [and if they are, it needs to be by formal amendment filed with the Institutional Review Board]. The confusion in all the deliberations is about what is in the CSR [Complete Study Report]. There is a long narrative, much more detailed than any published paper could ever be. And then there are Appendices. In the EMA system, the PROTOCOL is an appendix. The STATISTICAL ANALYSIS PLAN is an appendix. The SAMPLE CASE REPORT FORMS are an appendix. The compiled raw data [INDIVIDUAL PARTICIPANT DATA] is spread among several appendices. And finally, the actual CASE REPORT FORMS [CRF] filled out by the subjects or study personnel might be an appendix. So when people say CSR, it matters what appendices are actually included. Here’s what that email [some clarity?…] and the referenced documents said:
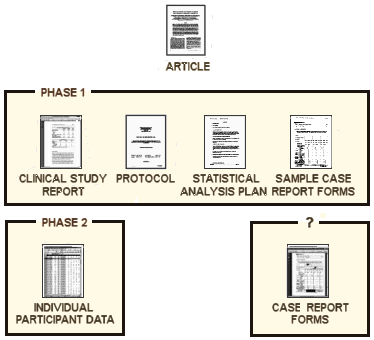
After a drug has completed the regulatory process, the documents in the phase 1 box above will be made available. The availability or conditions for availability of the various appendices with the raw data will be decided at a later date [phase 2].
Why I have perseverated [been hung up on] this point:
With Study 329, the CSR narrative was available and with that, one could tell something[s] was/were fishy. The PROTOCOL/STATISTICAL ANALYSIS PLAN became available along the way. In terms of efficacy, we needed the IPD to do the definitive analysis that we published in September [Restoring Study 329: efficacy and harms of paroxetine and imipramine in treatment of major depression in adolescence]. With only the article to go on, one could only speculate and suspect. It was over three years after the article was published that the CSR became available, and eleven years before the IPD was public. If the information in the phase 1 box above is available on every drug in a timely fashion, anyone can look to see if the reported article follows the protocol directed outcome variables using the pre·specified analytic techniques. That is a massive improvement over what we have now.
And if the phase 2 documents can be accessed when there is a just cause, the whole seedy racket of jury-rigging clinical trials that we’ve lived with for several decades will be quickly exposable. The issue of adverse effects is a special case in that the CRFs themselves may be required to determine harms.
Sometimes my eyes glaze over and I start skimming when you’re way down in the weeds of a study, but thinking out loud is often necessary and most beneficial to gaining insight, and I like watching you think out loud. Not all learning is fun, but it shouldn’t have to be.