When Dr. Bernard Carroll comments here, he often uses the term "hand waving" when describing some of the tricky maneuvers used in the clinical trial reports to smooth over shaky logic or rationalize absurdities. It’s a great term, I think originating in the world of stage magicians who use exaggerated gesticulations to distract your attention. My wife’s a figure skating fan, and there it’s called "hand dancing" – dramatic arm and hand gestures to cover up sloppy skating. After reading so many jury-rigged clinical trial reports, 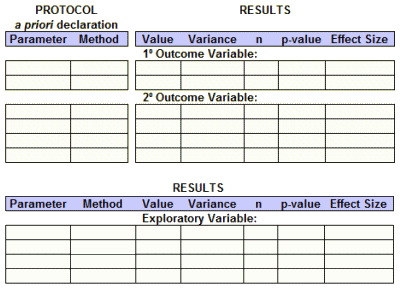 I’ve almost come to see the whole narrative as organized around a verbal version of these attempts at artifice, and find myself jotting down the essential pieces in a hastily sketched table on the back of a nearby envelope or piece of scrap-paper. So the basic efficacy table isn’t just a concept or a proposal. It’s an outgrowth of my experience. I guess the formula goes:
I’ve almost come to see the whole narrative as organized around a verbal version of these attempts at artifice, and find myself jotting down the essential pieces in a hastily sketched table on the back of a nearby envelope or piece of scrap-paper. So the basic efficacy table isn’t just a concept or a proposal. It’s an outgrowth of my experience. I guess the formula goes:
article narrative – bullshit = basic efficacy table
One thing the ghost-writers seem to count on is that most doctors look at the abstract, scan the graphs and tables, and move on. I used to see that as virtue – getting through so much material on a regular basis. In my doctor youth, I could do that. But no longer. If a Clinical Trial report or a review article is there to be read, it’s there to be read closely, pencil and envelope back at the ready. At least that’s true of the industry funded clinical trials of psychiatric drugs that I find myself reading these days.
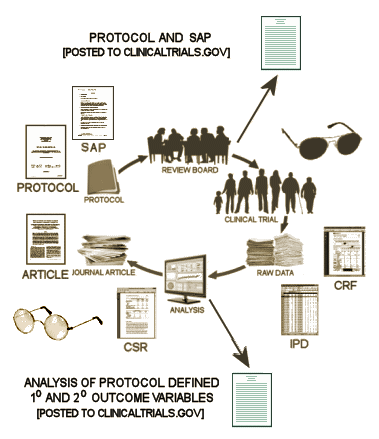
When I drew this diagram, it’s not how things are. It’s how I wish they would be. Step one is the approval of the study Protocol by the Institutional Review Board. At that point, by my reckoning, the trial should be registered [on ClinicalTrials.gov]. There’s no reason at all that the Protocol couldn’t be published at that point. It has been written down for the IRB. Why not make it a part of the registration process. That would mean that a bona fide copy of the a priori declarations would be available from the outset.
adapted from Table 1A inby Deborah A. Zarin, Tony Tse, Rebecca J. Williams, and Sarah CarrNew England Journal of Medicine. 2016 375[20]:1998-2004.
| Registration | ||
| When does information need to be submitted to or posted on ClinicalTrials.gov? | ||
| Submission: Within 21 days after enrollment of the first trial participant | ||
| Posting: Generally, within 30 days after submission. For ACTs of unapproved or uncleared devices, no earlier than FDA approval or clearance and not later than 30 days after FDA approval or clearance (i.e., “delayed posting”), unless a responsible party authorizes posting of submitted information prior to FDA approval or clearance | ||
| What information? | ||
| Descriptive information about the trial: e.g., brief title, study design, primary outcome measure information, studies an FDA-regulated device product, device product not approved or cleared by the FDA, post prior to FDA approval or clearance, and study completion date | ||
| Recruitment information: e.g., eligibility criteria, overall recruitment status, | ||
| Location and contact information: e.g., name of sponsor, facility information | ||
| Administrative data: e.g., secondary ID, human-subjects protection review board status | ||
| Results information reporting | ||
| When does information need to be submitted to or posted on ClinicalTrials.gov? | ||
| Submission: | ||
| Standard deadline: Within 12 months after the date of final data collection for the prespecified primary outcome measures (primary completion date) | ||
| Delayed submission with certification: May be delayed for up to 2 additional years (i.e., up to 3 years total after the primary completion date) for trials certified to be undergoing commercial product development for initial FDA marketing approval or clearance or approval or clearance for a new use | ||
| Submitting partial results: Deadlines are established for submitting results information for a secondary outcome measure or additional adverse information that has not been collected by the primary completion date | ||
| Extension request: After receiving and reviewing requests, NIH may extend deadlines for “good cause” | ||
| Posting: Within 30 days after submission | ||
| What information? | ||
| Participant flow: Information about the progress of participants through the trial by treatment group, including the number who started and completed the trial | ||
| Demographic and baseline characteristics: Demographic and baseline characteristics collected by treatment group or comparison group and for the entire population of participants in the trial, including age, sex and gender, race or ethnicity, and other measures that were assessed at baseline and are used in the analysis of the primary outcome measures | ||
| Outcomes and statistical analyses: Outcomes and statistical analyses for each primary and secondary outcome measure by treatment group or comparison group, including results of scientifically appropriate statistical analyses performed on these outcomes, if any. | ||
| Adverse event information: Tables of all anticipated and unanticipated serious adverse events and other adverse events that exceed a 5% frequency threshold within any group, including time frame (or specific period over which adverse event information was collected), adverse-event reporting description (if the adverse-event information collected in the clinical trial is collected on the basis of a different definition of adverse event or serious adverse event from that used in the final rule), collection approach (used for adverse events during the study: systematic or nonsystematic), table with the number and frequency of deaths due to any cause by treatment group or comparison group | ||
| Protocol and statistical analysis: Protocol and statistical analysis plan to be submitted at time of results information reporting (may optionally be submitted earlier) | ||
| Administrative data: Administrative information, including a point of contact to obtain more information about the posted summary results information | ||
First off, anything they do is a step forward. They’ve had the machinery available for two decades, and have done little with it. So they’re finally requiring registration for all the studies, and they pledge to keep up with it. An excellent start.
Initial Registration: They say that they want the submission of the trial registration within three weeks of the initial subject’s enrollment and posting on-line within the 30 days after submission. Of course I’d prefer our "before the study starts" timing, but within the first two months will do. The point is to get it registered before they can look at the results and modify the Protocol – and two months is early enough for me. As for what’s to be posted, they don’t require posting the whole Protocol. That’s a disappointment. I’d prefer anchoring the outcome parameter at the beginning. But at least they do require declaration of the Primary Outcome Variables with registration.
Posting the Results: This has traditionally been the most ignored requirement. They say: "Outcomes and statistical analyses for each primary and secondary outcome measure by treatment group or comparison group, including results of scientifically appropriate statistical analyses performed on these outcomes, if any" and add in "Protocol and statistical analysis plan to be submitted at time of results information reporting (may optionally be submitted earlier)." And I say A+! With that information, I could fill out my entire basic efficacy table, The only thing they left out was the Effect Size and there would be ample information to do that calculation.
And for timing on the Results? I’d have to say "barely passing," if that. "Within 12 months after the date of final data collection for the prespecified primary outcome measures (primary completion date)" and "May be delayed for up to 2 additional years (i.e., up to 3 years total after the primary completion date) for trials certified to be undergoing commercial product development for initial FDA marketing approval or clearance or approval or clearance for a new use." That’s a disappointment, and I can’t see any reason for it. The results are just what they are – they’re the results of the prespecified variables analyzed in the prespecified way. Who needs time for that? But I’ll have to admit that if they were to actually to follow these standards, the improvement would still be dramatic, probably satisfy most of us. With new drugs or new indications, they’d still be early in the drug’s patent life.
-
the a priori declared primary and secondary outcome variables
-
the prospectively defined statistical analysis plan
-
the values, variance, and effect size for those specific parameters
This is well put, Dr. Mickey. The Final Rule that came out last fall is a step in the right direction. We share deep reservations about the potential 3-year post-completion delay in posting the true protocol and plan of analysis for the prespecified outcome measures. In our petition we pointed out that the FDA already gets that information. Nobody has given us a good reason why FDA cannot monitor what does or does not get posted on ClinicalTrials.gov. If these 2 agencies could synchronize and harmonize their information then transparency would be served.
”
•the a priori declared primary and secondary outcome variables
•the prospectively defined statistical analysis plan
•the values, variance, and effect size for those specific parameters ”
We have 19 million reasons to perform scientifically valid research:
http://www.medicaldaily.com/x-files-clinical-trials-human-experimental-subjects-372422
And the consequences of not following the guidelines are ? ? ?
Something I’ve been wondering, though I haven’t searched on it. Maybe you know. Who determine what adverse events or reaction are mild vs serious?
While looking at various pharmaceutical products’ categories for negative events, my idea of what I would consider serious is never the same as what the black words on the white paper product inserts tell me. Is there a standardization of these categories? When deciding whether or not to take a medicine, people tend to look at the serious reactions and skip the rest. What if the rest are pretty serious in the layman’s eyes, but have been deliberately relegated to a less concerning list to avoid notice?
Not trying to exonerate psychiatry by pointing fingers elsewhere but the hand-waving seems to be everywhere.
This from Pharmalot (article behind paywall)
“Over the past few years, billions of dollars have been spent on new hepatitis C medicines because they could eliminate the virus in 90 percent or more of patients. But a new report finds that hundreds of cases of liver failure were associated with the drugs, and the authors suggest that regulators may have been too quick to embrace the treatments as a panacea.”
http://www.statnews.com/pharmalot/2017/01/25/hepatitis-safety-gilead-fda/
Having the basic efficacy table is an excellent first step.
But what happens next? Would that information actually change anything in terms of prescribing patterns? The BBC reported in 2014 that the 10 largest drug companies spent 100 billion dollars in marketing. Assuming that the remaining drug companies spent an equivalent amount, that means that the drug companies spent half a billion dollars a day on marketing.
That number makes my brain melt. Half a billion dollars a day on telling physicians and the public to prescribe / take certain pills. How will the data from efficacy tables be an effective barrier to that cannonade of money?
Eric,
Good point, but I think it would make a difference. In the antipsychiatry literature, psychiatrists are presented as “bought” men or “made” men – super-vulnerable to false advertising. That’s what the KOLs have done for our specialty and why they deserve a lower circle in Dante’s Inferno for rationalizing their Conflicts of Interest and “signing on” to. questionable or exaggerated studies. If there’s a readily available conduit to well presented information, a lot of that will change, all by itself….
As I once was considered an astute LISW on the adult psych floor I am again putting my two cents in. There were phsycians that I held in high regard and then there were the others.
Since I also was invited to be in an academic journal club with members who went on to testify in front of Congress and was involved in many psychoanalytic seminars I probably have more than two cents to throw in.Yes, anti psychiatry literature does not present your profession well for several legitimate reasons.It is a mess and a con of worms on all sides, academic, research, clinical act.
All of you here that are medical professionals at least have part of the blinders off and a re willing to above and beyond on thinking about research. And I thank you.
However, has a professional and as a former patient who was thrown in during a time of abject stresses on multiple levels I can see clearly where the anti folks are coming from.
Besides the undoing and getting of good research and dialogue of what really works and what really doesn’t – i.e. swan catheters – I remember those days.
There needs to be some true work on apology. Leon Uris in one of his books talked about apology. It was a page and half of true brilliance about the responsibility of a vase broken by a gust of wind. Whether it was the wind’s fault or not the vase keeper who put in the drafty hallway while the owner was away needs to acknowledge responsibility though there are buts. The keeper of the vase also has to acknowledge I am sorry for the loss even though the keeper may have thought it was a terrible replication of work of art. And then the keeper needs to make amends for the loss even though both parties know the vase can never be perfectly replaced.
There is a reason the Catholic Church made confession a sacrament and the use of it in the best way during the times of the great Irish monks and communities.
Harm was done to many people. All the words in the world can not obsatafucate sp? that fact.
It looks as if Big Pharma or Research Academia may never unless legally forced admit to any type of wrong doing. I am guess they will follow the Big Tobacco path going into underdeveloped and unaware counties.
I would posit that some sort of professional apology be made to the patient community. Harm was done in a profession that once held “Do No Harm” in great respect. Many medical schools did drop that phrase – my father gave a medical graduation speech and that was his entire theme – the importance of do no harm as a ethical tenant in the medical community.
One cannot change this election but one can and with others do the right thing.
I am not sure this comment will fit this specific thread accurately, but having just read in the recent issue of Clinical Psychiatry News and the front page story almost glorifying the agenda of pushing to make MDMA a “medication” for treating PTSD, as they write in the underbanner, “the love drug explored as PTSD treatment”, well, what the hell??!!
I write about it here after reading Dr N’s above comment in the thread, hopefully accurately highlighting the motivation to mention it:
“Good point, but I think it would make a difference. In the antipsychiatry literature, psychiatrists are presented as “bought” men or “made” men – super-vulnerable to false advertising. That’s what the KOLs have done for our specialty and why they deserve a lower circle in Dante’s Inferno for rationalizing their Conflicts of Interest and “signing on” to. questionable or exaggerated studies. If there’s a readily available conduit to well presented information, a lot of that will change, all by itself….”
I know Dr N has addressed Ketamine in past posts, but, this push to legitimize drugs that are responsibly and appropriately listed as Controlled 1 substances, it is beyond frightening for me.
Just hoping this opinion legitimately adds to this discussion…
Oh, the article:
http://www.mdedge.com/clinicalpsychiatrynews/article/126720/ptsd/mdma-love-drug-makes-therapeutic-comeback
Ketamine will probably be replaced by its less toxic active metabolite, which relieves depression not through NMDA receptor mechanisms but AMPA mediated pathways. I’m not sold yet, but I find the research interesting.
The work with MDMA assisted psychotherapy is actually not new but a reiteration of a lot of work that was done up until the mid-eighties. Despite the club drug reputation of MDMA, I support this because anything that cuts through resistance is going to save psychotherapy. It’s simply not going to be able to go on and on Woody Allen style in the age of rationing and capitation. Mithofer’s (?sp) pilot experiments at U South Carolina on veterans looks promising. I’m in agreement with Julie Holland, M.D. and others that this is important work because the drugs we have for PTSD are not good.
I don’t have a problem with well-designed work in this area. Frankly, the widespread acceptance of medical marijuana is a lot less evidence based than these studies. This kind of work seems a lot more promising than another me-too SSRI, which has obviously reached a biological and financial dead end.
This kind of thing bothers me a lot more than MDMA or ketamine research on adults….pot research on kids…apparently he is serious:
http://www.mdedge.com/clinicalpsychiatrynews/article/130188/pediatrics/marijuana-calms-children-autism