Posted on Monday 10 October 2016
In the days before television and other devices, some satirists used the comic strip for their commentaries – none more effectively than Walt Kelly who drew Pogo. Pogo possum and Kelly’s other anthropomorphized denizens of the Okefenokee Swamp delighted us with political satire and commentary on the human condition for a quarter century. There was only one character, Pup Dog, who remained on all fours as a playful puppy and only said "wurf." But there was a period, where he said, "Poltergeists make up the principal type of spontaneous material manifestation" – a nonsense Tautology that translates to "ghosts are ghosts."

To be honest, these short term clinical trials don’t really need much writing by anyone at all. Some simple tables would do just fine: a list of the primary and secondary outcome variable defined before the study started with their results; the subjects’ demographics and drop-out rates; some comparative tables of adverse events; and a special table for severe adverse events. Write a few sentences at the end summarizing the conclusions. How their assertion that "In fact, the contributions and expertise of medical writers working with authors is associated with more complete reporting of the results of clinical trials and higher quality content" fits into that is unclear [and highly unlikely]. The notion of the hired-on ghostwriters and the hired-on KOLs working hand in hand to produce a value-added article is a fabulist scenario. Neither has seen the data or run the numbers, so the whole picture they’re painting in their response to Matheson is a fantasy. We have a few opened windows into the ghostwriting process via subpoenaed internal emails and documents from various lawsuits. The general pattern is for the ghostwriter to get some kind of summary document from the Sponsor to turn into a first draft. After it’s tweaked by the Sponsor, it’s cleared for the "authors" to take a look at – hardly the royal road to "higher quality content."
"ISMPP’s long-standing position is that “ghostwriting” – defined by Laine and Mulrow in 2005 as individuals who wrote the paper but are not acknowledged’ – is an unacceptable practice…"
"ISMPP fully supports the role of professional medical writers and the complete and transparent disclosure of their involvement in medical publications and the source of their funding."

"… My analysis shows, however, that modest transparency and small print disclosure, while better than nothing, paradoxically provides an apparatus for subtle misattribution, concealment and spin. My article discusses this as a cultural problem within medicine for which many stakeholders are responsible. It is important not to lay the blame at industry’s door alone. As Stretton rightly points out, it is journals that make authorship rules, and decide how disclosures should be presented to their readers. Table 2 of my article provides some useful suggestions to the editorial community."
"More importantly, however, while I applaud Stretton’s and Weigel’s commitment to transparency, I remind them that the publications trade remains unacceptably secretive. Numerous articles are produced for journal publication every year, scientific platforms and publications plans are developed, academics are recruited to participate as authors, trade writers draft manuscripts, and substantial payments pass from corporations to agencies, but little of this activity is in the public domain. Indeed, the process has been characterized as ‘ghost management’. Commercial secrecy has no place in an open scholarly discourse…"
This affirmation of continued commercial secrecy on these aspects of medical journal literature – made directly to the medical community itself, in a leading medical journal – will be of broad ethical and policy concern. The joint statement ends by claiming the trade has been “working towards” transparency and disclosure for 15 years. There is clearly a long road yet to travel…

 business and our
business and our 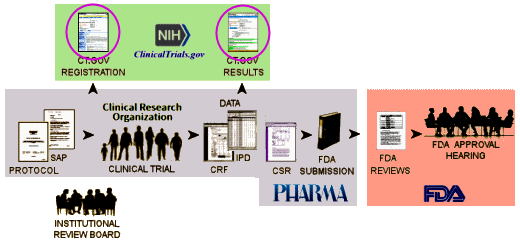
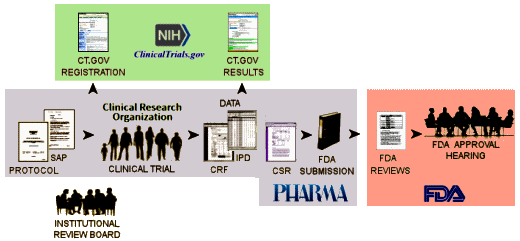
 But that’s not what the NIH/ClinicalTrials.gov/FDA did [see
But that’s not what the NIH/ClinicalTrials.gov/FDA did [see