In the course of life, I had a hand in the later raising of an orphan, and I knew nothing of an orphan mentality. She hadn’t been abused, but she had been chronically deprived for much of her life. To my surprise, she was never satisfied. She had no concept of "enough" – so she always felt disappointed and was only comfortable in a somewhat deprived state. I had to learn that my natural inclination to be on the indulgent side [making up for the past] only made her sick, and that the right course of action was to provide only the "right amount" and accept her chronic state of disappointment as her necessary comfort zone. "Too much" was just as disastrous as "not enough." I’m pleased to report that it worked out in the end, but the path was slow and had more than its share of bumps. While it’s a trivial adage, "don’t go to the grocery store hungry" seems to apply. Substitute the word "starved," and you’re close to understanding the orphan dilemma.
EMA adopts landmark policy to take effect on 1 January 2015
Press release
October 2, 2014
The European Medicines Agency [EMA] has decided to publish the clinical reports that underpin the decision-making on medicines. Following extensive consultations held by the Agency with patients, healthcare professionals, academia, industry and other European entities over the past 18 months, the EMA Management Board unanimously adopted the new policy at its meeting on 2 October 2014. The policy will enter into force on 1 January 2015. It will apply to clinical reports contained in all applications for centralised marketing authorisations submitted after that date. The reports will be released as soon as a decision on the application has been taken.
“The adoption of this policy sets a new standard for transparency in public health and pharmaceutical research and development,” said Guido Rasi, EMA Executive Director. “This unprecedented level of access to clinical reports will benefit patients, healthcare professionals, academia and industry.”
The new EMA policy will serve as a useful complementary tool ahead of the implementation of the new EU Clinical Trials Regulation that will come into force not before May 2016. EMA expects the new policy to increase trust in its regulatory work as it will allow the general public to better understand the Agency’s decision-making. In addition, academics and researchers will be able to re-assess data sets. The publication of clinical reports will also help to avoid duplication of clinical trials, foster innovation and encourage development of new medicines.
According to the policy’s terms of use, the public can either browse or search the data on screen, or download, print and save the information. The reports cannot be used for commercial purposes. In general, the clinical reports do not contain commercially confidential information. Information that, in limited instances, may be considered commercially confidential will be redacted. The redaction will be made in accordance with principles outlined in the policy’s annexes. The decision on such redactions lies with the Agency.
The policy will be implemented in phases. The first phase starts on 1 January 2015. Once a medicine has received a marketing authorisation, EMA will publish the clinical reports supporting applications for authorisation of medicines submitted after the policy’s entry into force. For line extensions and extensions of indications of already approved medicines, the Agency will give access to clinical reports for applications submitted as of 1 July 2015 after a decision has been taken.
In future, EMA plans to also make available individual patient data. To address the various legal and technical issues linked with the access to patient data, the Agency will first consult patients, healthcare professionals, academia and industry. It is critically important for EMA that the privacy of patients is adequately protected before their data are released.
The policy does not replace the existing EMA policy on access to documents. It will be reviewed in June 2016 at the latest.
The decision of the EMA has been long awaited and is still incomplete. When it showed up yesterday, I scanned it and some of the reactions – then let it sit for a while. I realized that in this case, I’m in the orphan class ["
been down so long it looks like up to me"]. And reading
worry…, it’s easy to see that I’m primed to be disappointed. It’s not hard to see paranoia in others, but not nearly so simple in the mirror. Inserting some time is a good antidote. So if I can be allowed to revive my monotonous graphics:

There are four documents generated from a Clinical Trial: a Protocol, the CRFs [Case Report Forms], the IPD [Individual Participant Data], the CSR [Clinical Study Report]. Oh yeah, I guess we could add the published [or maybe unpublished] journal article, bringing the total to five documents [see it matters…]. In the absence of outright fraud, three of them come from "behind the blind," in other words, they are what the investigators [and sponsors] see when the "blind is broken." If you have those three things, the playing field is level. Someone evaluating the study from afar is in the same boat as those who did it. We’re talking about a gajillion pages [every piece of paper from every visit by every subject, the CRFs, AND the tables compiled into the IPD of everything that can be objectified and tabulated]. It’s an overwhelming stack of stuff.
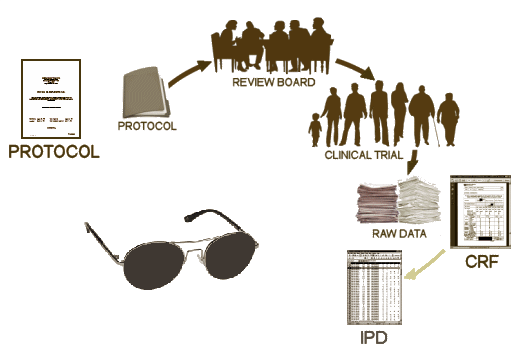
After the blind is broken and the data is at hand, it is collated, analyzed, and turned into the CSR – a manageable report for regulators that will later be simplified further to become a journal article [or not]. While there are plenty of computers whirring at this point in the process, there are real live people with real live motives also in the mix, and this is where the problems have come from – beyond the blind:
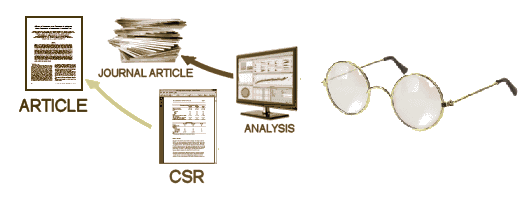
What the orphan in me wants is all of it. I want the Protocol, the CRFs, and the IPD. I want to to be level with the investigators/sponsors. I’m understandably "starved." The CSR will contain the Protocol and in some cases, the IPD as Appendices, but that last part isn’t guaranteed, and I doubt it will continue under this policy. The raw CRFs won’t be in the CSR. So where does that leave us? The downside is that the CSR is prepared by those pesky, motivated humans. So the possibility of sleight of hand is there as it is with the messy journal articles we’ve lived with for too long. And, they get to pick what’s in them. On the other hand, it puts us on the same level as the regulators who have the ultimate power – saying "No" to approving the drug. And that first paragraph up there has a key sentence, "The reports will be released as soon as a decision on the application has been taken." And this promise sounds pretty good too, "In future, EMA plans to also make available individual patient data." Not guaranteed, but at least an acknowledgement of the need. The piece that’s missing is the CRFs, necessary for a thorough vetting of the Adverse Events.So does it pass the Rolling Stones test:
You can’t always get what you want
You can’t always get what you want
You can’t always get what you want
But if you try sometimes
Well you just might find
You get what you need
Some may see this as a
Neville Chamberlain conclusion, but on reflection, it seems to me that the European Medicines Agency is giving us the same thing they get for themselves, and we can’t ask for much more than that. The one thing missing is a retrospectoscope. There are lots of previously approved drugs we need to know about, because, as Ben Goldacre says, they’re still in use. That’s an area where we need to challenge the EMA. But the other part, the
IPDs and the
CRFs which are needed to vet questionable studies is probably a fight that needs to be taken directly to the drug manuacturers through the data portals they are creating. It seems naive to ask the EMA to take on the responsibility of getting us something they don’t even have themselves. I give them a B+ [but reserve the right to change my mind]…
UPDATE:
This from Ben Goldacre:
“Firstly, the EMA records are woefully incomplete for informed decision making: EMA only holds CSRs for a small proportion of all the trials done on all the medicines we use today. We need a radical overhaul giving retrospective transparency on all CSRs from industry, and clear transparency on methods and results for all trials done by academics. Secondly, this policy does nothing to move forward on the safe sharing of individual patient data – whilst respecting patient privacy – which was promised by EMA in 2012. Lastly, there are serious concerns around the redactions process. For this, we can only go on recent performance, which is not encouraging. EMA reached an agreement this year with AbbVie to censor information on protocol changes from the public release of a CSR. Protocol changes in a trial are precisely the kind of information that researchers need, to make an informed decision about whether that trial was a “fair test” of the treatment. It is hard to see how it is justifiable to hide protocol changes, in a trial from eight years ago, on over-riding grounds of commercial confidentiality.”

A step in the right direction but now I’m beginning to wonder if transparency isn’t enough. (Necessary but not sufficient.) I think you could have archangels doing RCTs under current protocols and still produce garbage because of the high placebo response. See my Feynman speech/rat maze analogy:
http://1boringoldman.com/index.php/2014/09/24/a-lot-riding-on/#comments
The next step is to start testing the drugs on sicker inpatients (or at least more supervised patients) using a far more stringent criteria than DSM5 MDD. Noise reduction. Anything with a placebo response of 40 pct is garbage. The signal to noise ratio in modern SSRI studies reminds me of a cheap transistor radio made in 1961.
James,
I absolutely agree. RCTs are intrinsically flawed. I think David Healy has a solid take on that point. But I decided that my focus is going to be on the most immediate problem, simple honesty in reporting. My sense of things is that RCTs aren’t going to go away, at least in the short term, because they intuitively nake sense. You have to do some serious thinking to understand why they are flawed. Honesty, on the other hand, is easy to see and an immediate need. RCTs will do for efficacy. But Adverse Events are another matter. Again, David’s idea of RxISK is a good one, though I’m not sure that there’s not an official, binding, long term format that needs thinking about.
PS I love the Cargo Cult story and the rats walking on sand corollary. Great stuff…
It’s just so disheartening to think we knew better how to isolate the independent variable in 1954 than we do in 2014…60 years of bad habits are not going to be erased even with ethical reforms…so many giant steps are going to have to be taken against so much institutional resistance that proclaims that sand in the rat mazes doesn’t matter.
“It will apply to clinical reports contained in all applications for centralised marketing authorisations submitted after [1 January 2015].”
If there are independent parties analyzing the clinical reports, this will be helpful to doctor and patients 5-10 years from now, after the pipeline produces products. It’s something, I guess, but I’m with Ben Goldacre — the records need to be released retrospectively, those are the ones germane to the drugs people are taking now.
Alto,
I agree. But from what Ben says, the EMA may not have them. The part you bring up is vital, but I think that fight might have to go to the drug companies. Arrgh!
I think every study should be recorded with video and sound for both subjects and those doing the testing, and the whole kit and kaboodle should be published and open to review.
Patient selection is key but DSM makes that impossible since MDD now includes so many marginal conditions that used to be adjustment disorders.
Research has to follow NIMH and discard DSM MDD to be worth a damn.
I would also get back to adding an MMPI for additional verification and weeding out test subjects with overreporting/underreporting issues and possible substance issues.
I know it’s impossible to weed out all the noise, but they could try a lot more.
Before good studies are done on MDD drugs, the study on how to get the placebo response rate below 20-25% needs to be done.