Well. it seems far, far away from now. It was the 1960s, and the notion that someone on a medical faculty might have financial ties to a pharmaceutical company was
unheard of. Coming this way into the 1970s to a psychiatry residency and faculty appointment – the same
unheard of. I wrote about my first encounter with a drug company sponsored event in a training program a few years back [
repressed memories…]. It was 1983-ish. There was a new chairman, and it was a new day [post 1980 DSM-III]. The new chairman wanted me to start a Grand Rounds series with outside speakers. We had no funds for such a thing, but he said not-to-worry, he’d take care of it. Here’s what I wrote four years ago looking back:
At one of the first Grand Rounds, he had someone from the faculty of the University of Georgia. I thought it was a great idea. There hadn’t been much dialog between Emory [Atlanta] and the Medical College of Georgia [Augusta], and this seemed like a good way for us to get together. The lecturer who came had pretty slides, but as he talked, it began to sound like a sales pitch for Mellaril [Sandoz] rather than a scientific presentation. I was disappointed appalled. I thought the new Chairman would be embarrassed, but he didn’t seem to be. I left Emory at the end of that year. It was mutual, I think. I just didn’t fit anymore. So I didn’t think about that presentation for a decade. The speaker that day was Dr. Richard Borison, who became Chairman of Psychiatry at the Medical College of Georgia. He was a drug researcher, and I started seeing his name on journal articles, always drug studies…
Over a decade later, Chairman Richard Borison’s Clinical Research Center was "busted" and he was sent off to prison for embezzling millions from the University of Georgia [Drug Money Patients Worsened; Little Oversight Provided]. But I’m getting ahead of myself. At the time, I was both appalled at having a drug company sponsored infomercial for a Grand Rounds and also understood why the new chairman did it. We were flat broke. Money for training had just dried up and we were literally running on flumes. All of this happened shortly after the DSM-III appeared. Others more in-the-know than I assure me that industry had nothing to do with the DSM-III Revolution – that it was later opportunistism. But I still wonder from time to time.
In the years that followed, I was no longer directly involved, but even from across town, it was obvious that they were in the land of milk and honey through Emory’s Nemeroff years [1991-2009], and there was nothing subtle about the industry sponsorship. It was on display – the norm. For a long time, industry ties weren’t apparent in journal cites. Then they were declared in COI statements [and they were everywhere!]. So I find reading this Editorial in the British Medical Journal that goes even further very satisfying:
Editorial
by Mabel Chew, Catherine Brizzell, Kamran Abbasi, And Fiona Godlee
British Medical Journal 2014 349:g7197.
Zero tolerance on education articles with financial links to industry
The BMJ was one of the first medical journals to seek declarations of competing interests from authors. Our focus is on financial competing interests as we believe these to be the most identifiable. We do, however, understand that competing interests come in many forms and we also routinely ask authors to declare relevant non-financial competing interests. The governing principle has been that transparency is a panacea. We placed faith in this principle, but mounting experience and evidence tell us that we were only half right. Transparency remains essential, but it isn’t sufficient to eliminate bias or perception of bias.
We believe this risk of bias is particularly important for clinical educational articles that are designed to guide patient care, when authors’ biases may be less visible to general medical readers. For some years we have sought to minimise as well as declare competing interests for these articles. Recently we introduced more active management of competing interests, requiring authors to complete a more detailed declaration and excluding authors with close ties. Now we have decided to go a step further, as heralded three years ago. From next year our clinical education articles will be authored by experts without financial ties to industry [box]. By industry we mean companies producing drugs, devices, or tests; medical education companies; or other companies with an interest in the topic of the article. We are phasing in this policy to start with editorials, clinical reviews, and most practice series. We hope that by the end of 2016, this will have extended to the rest of our education section: our specialist state of the art reviews and diagnostics and therapeutics series.
-
Competing interest definitions and process for The BMJ’s editorials and education articles [including clinical reviews, practice articles, and state of the art reviews]
-
“A conflict of interest arises when a person has a personal or organisational interest that may influence or appear to influence the work they are doing. Usually this is a financial interest, but it may also be non-financial.”
-
We ask authors to declare interests in the 36 months before the declaration and those known to be going to occur during the next 12 months
-
Authors are asked to complete a form, available at www.bmj.com/sites/default/files/attachments/resources/2011/07/current-bmj-education-coi-formfinal-1.doc. For unsolicited articles, we also ask who prompted submission and whether professional writers contributed
-
Each author’s declaration is carefully assessed by the handling editor, and may be discussed at a regular editors’ meeting, to ensure our decisions are consistently and fairly applied by the editorial team.
-
We have started publishing authors’ competing interests forms alongside the articles, and advise authors of this when they send their forms. We plan to do so for all editorials and education articles
-
From 2015, we will roll out a policy of editorials and clinical education articles authored by experts without financial ties to industry [companies producing pharmaceuticals, devices, or tests; medical education companies; or other companies with an interest in the article topic]
Shift in culture
Why are we doing this? The first reason is that making clinical decisions based on information biased by commercial interests can cause harm, as happened with cardiotoxicity from rosiglitazone and rofecoxib and continues to happen with hydroxyethyl starch. We also believe that the educational content we publish will have more impact if readers can trust it. We know that readers consider research papers written by authors with declared financial links to industry to be less important, relevant, rigorous, and believable; they are also less willing to prescribe drugs evaluated in such papers. Finally, we want to encourage a shift in the culture of medicine. We think that we can help to do this by promoting authors without financial ties to industry and offering them appropriate prominence and visibility.
Financial competing interests are endemic to the culture of medicine and are rarely driven by malign motives or actions. The mechanisms of influence are diverse. An author of a review article might be an advisory board member for companies selling drugs for that condition, a commentator might have received honorariums from industry for lectures on the topic, or an editorialist on a disease might be a patent holder for one of its diagnostic tests. Psychological research suggests that biases may operate subconsciously. Our decisions not to proceed with an article or an author are not made lightly. Nor are they intended to pass judgment on an author’s integrity. However, we cannot ignore the mounting evidence of systematic attempts by commercial interests to corrupt the literature and influence clinical decisions. Internal company documents revealed during litigation expose practices aimed at influencing clinicians such as funding medical meetings, dinners, studies, and articles. Many clinical practice guidelines are little more than industry marketing tools because of the financial competing interests of their authors and sponsors…

During all those years, I didn’t like the prominence of pharmaceutical influence everywhere, but may have mistakenly thought it was like the Grand Rounds I mentioned – desperation financing. It was that, but so much more. It was ghost writing, jury-rigged Clinical Trial reporting, Speaker’s Bureaus, payola, etc. It wasn’t just money flowing to institutions, it was going directly into the pockets of academic physicians and other KOLs. Industry was able to buy prominent doctors with a small fraction of the money they made by having them on board. I would now say that my little Star Wars graphic isn’t that far off the mark. It was something of a Galactic Empire and it’s still there. I think of psychiatry as the worst offender, but that may only be because I know more about what went on. But when I look at my television set or hear the MRI machines whirring in the background when I go in a hospital, I suspect it’s all over Medicine proper too [it being outside influences pushing the profitability of Medicine wherever it can be pushed in whatever way it can be pushed].
And we can’t operate on a bad apples theory. Too many physicians in high places were [are] on board [just look at PROPUBLICA’s Dollars for Docs or the Sunshine Act site]. We have to swallow our disillusionment and assume that enforcable [and enforced] stops will be required to keep it from continuing and/or recurring [same it]. We have now a whole generation of physicians who’ve grown up in this climate, with too many participating, and they need special attention. So it’s not just a matter of putting restraints on industry, it’s equally important to focus on physician collusion and respond decisively.

And as for Dr. Fiona Godlee and her editorial staff at the BMJ – center of right-thinking for a long time: she’s testifying in Parliament, leading in the fight for the Tamiflu data, setting policies like this one, doing everything in her power to lead us out of the wilderness and
"encourage a shift in the culture of Medicine." Does this kind of activism count in Stockholm? Do journal editors qualify for Knighthood? Is there some way to recognize her persistence and her savvy in knowing what rocks to look under? Could she be cloned? Fiona Godlee is a definite "keeper."


 -n
-n k
k r
r z
z

 m
m
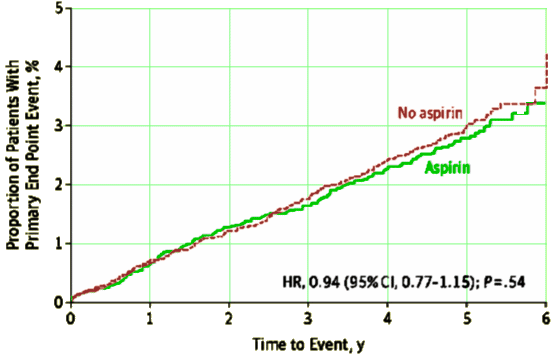
 My point isn’t about baby aspirin. I don’t take them myself, but if I had exertional chest pain, I might. If I ever have an oppressive substernal chest pain radiating down my left arm, I bet I’ll take Aspirin first, then call 911. My point is about the use of statistics in medicine in general. I’m moderately savvy as doctors go about statistics – a numbers guy by nature who likes quantification in almost any flavor. But statistical predictions feel out of hand to me. My specialty, psychiatry, has gone through a very long period where small differences have often been magnified to an outrageous degree and mere statistical significance has been presented as a surrogate for true clinical relevance. It’s not.
My point isn’t about baby aspirin. I don’t take them myself, but if I had exertional chest pain, I might. If I ever have an oppressive substernal chest pain radiating down my left arm, I bet I’ll take Aspirin first, then call 911. My point is about the use of statistics in medicine in general. I’m moderately savvy as doctors go about statistics – a numbers guy by nature who likes quantification in almost any flavor. But statistical predictions feel out of hand to me. My specialty, psychiatry, has gone through a very long period where small differences have often been magnified to an outrageous degree and mere statistical significance has been presented as a surrogate for true clinical relevance. It’s not. 
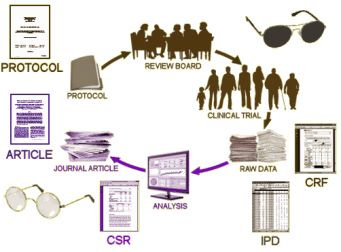
 We all know, that’s not what happens. The results are turned over to the corporate sponsor who analyze the results and use medical writers to produce the article. In many cases, the physicians and other scientists on the Author By-Line are primarily a ticket into a peer-reviewed journal, and have little if any input into the actual production of the article. And since the reader of the article can’t see any of the process that goes into the creation of what they’re reading, the data can be subtly manipulated to accentuate the positive and eliminate the negative. It not only can happen, it has happened in epidemic proportions – yielding enormous profits and more importantly has resulted in doctors prescribing many ineffective or sometimes harmful medications. The time for simply decrying this state of affairs has long passed. It’s time to put a stop to it, but that’s proving to be a very thorny task.
We all know, that’s not what happens. The results are turned over to the corporate sponsor who analyze the results and use medical writers to produce the article. In many cases, the physicians and other scientists on the Author By-Line are primarily a ticket into a peer-reviewed journal, and have little if any input into the actual production of the article. And since the reader of the article can’t see any of the process that goes into the creation of what they’re reading, the data can be subtly manipulated to accentuate the positive and eliminate the negative. It not only can happen, it has happened in epidemic proportions – yielding enormous profits and more importantly has resulted in doctors prescribing many ineffective or sometimes harmful medications. The time for simply decrying this state of affairs has long passed. It’s time to put a stop to it, but that’s proving to be a very thorny task. This history has been written by many well-meaning congressmen, reformers, and agencies, but they haven’t stayed at it. Industry is always at it. So the crises pass and drug companies just keep on doing what they’ve been doing as soon as the dust settles. It’s time for a new clamor – enforcement and ongoing monitoring. Without those two things, it’s all just more empty promises…
This history has been written by many well-meaning congressmen, reformers, and agencies, but they haven’t stayed at it. Industry is always at it. So the crises pass and drug companies just keep on doing what they’ve been doing as soon as the dust settles. It’s time for a new clamor – enforcement and ongoing monitoring. Without those two things, it’s all just more empty promises…