

Posted on Thursday 29 September 2016
[see also clinical trials – an act of congress!…]
In the course of a Clinical Trial on an FDA regulated drug, the Sponsor submits information to three distinct entities – each with a different purpose:
-
NIH [clinicaltrials.gov]: …clinicaltrials.gov is intended to be the public interface for the clinical trial, The first submission [registration] describes the trial and lays out how it is to be conducted and should be submitted after the study has been approved by the Institutional Review Board and before the study begins. It is a proxy for the a priori Protocol identifying the Primary and Secondary Outcome Variables and how they will be analyzed – a basic element of the RCT process. At the end of the clinical trial proper with the breaking of the blind, there is a second submission to clinicaltrials.gov [results database] giving the results of the analysis of the Primary and Secondary Outcome Variables. clinicaltrials.gov hasn’t had the desired impact, not because it wasn’t a good idea or well designed. People just ignored it. Trials have not been registered prior to starting the study. The results database has rarely been populated on time, if at all. The only consequence for ignoring it, even when it’s required by law, is being listed in one of many articles documenting that it’s being ignored.
-
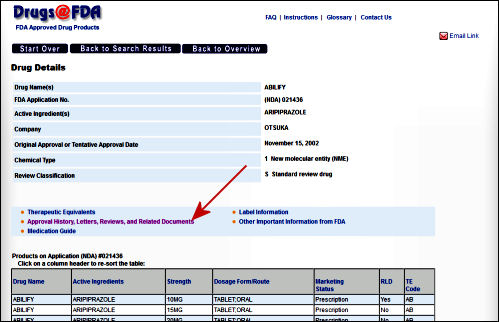
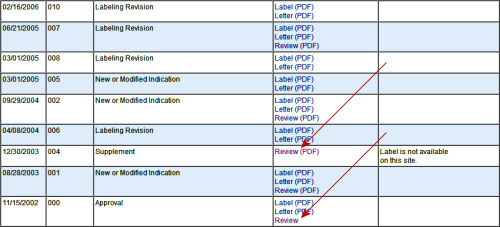
FDA: …The Sponsor’s submissions to the FDA re formal requests for a New Drug Approval, an approval for a new indication, an application for the pediatric extension of Exclusivity. Those submissions include the results of multiple trials [see Evidence of Clinical Effectiveness and Data Requirements For an NDA]. The Sponsor’s FDA Submission is not publicly available and the results of the FDA’s analyses are not published promptly – added much later for selected studies on Drugs@FDA. Those not posted can be requested via an FOIA request. The FDA’s decisions are yes/no and what’s published are the mutually negotiated package inserts whicg are collected yearly in the PDR [Physician’s Desk Reference].
-
Medical Journal: …articles in peer reviewed academic medical journals have been the traditional source of physicians for medical information for well over a century. Academic Medical Journals are independent and self-regulated. In the case of these clinical trial reports, the only information the editors and peer reviewers have is what’s submiited by the trial sponsor. No one these days think that the data was analyzed or the paper was written by the academic authors on the by-line. The analysis was done by the sponsor and the paper written by a professional medical writer from a prepared summary provided by the sponsor. The listed non-industry authors are involved in that to a greater or lesser extent. Over time, the purpose of the publications has essentially become a powerful advertising tool for marketing the drug – often inflating efficacy and minimizing harms.
So in spite of the fact that there should be four versions of the data results for a clinical trial, from where we sit, there’s only one. The clinicaltrials.gov version is usually empty. The IPD [Individual participant data] and the CRFs [Clinical Report Forms] are locked away in the sponsor’s filing cabinet/computer. The only other access is through the FDA who is sworn to secrecy. And so all we can see is the published article. Not even the ghost-writers, the listed academic authors, the journal editors, or the peer reviewers have access to the actual data and unfiltered results in the study.
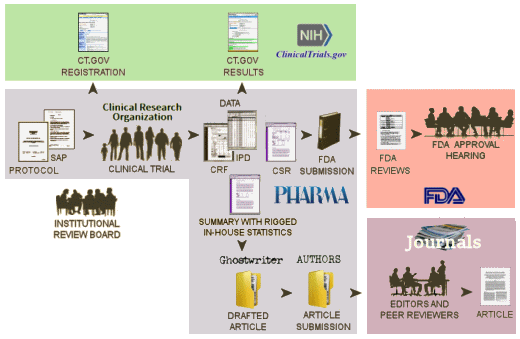
Notice the bifurcation in the pathways followed for the FDA Submission and the Article Submission. Add in the fact that both clinicaltrials.gov submissions are either misused or ignored. So while we appreciate the recent initiatives by the NIH and the FDA to do something about all of this, what they propose is not enough! In the current system in the graphic above or the system as proposed by their initiative, the only entity with eyes-on access to the raw data [the FDA] surveys neither the information on clinicaltrials.gov nor that published in journal article for accuracy.
So that’s why we’re urging you to sign our petition for Congressional action mandating that the FDA and NIH take an active role and use these already-in-place tools to put some teeth into ending the scientific disgrace of our clinical trial reporting! That’s what these agencies are for!

[click the ostriches to see and or sign the petition]
Comments Off on please sign the petition!…

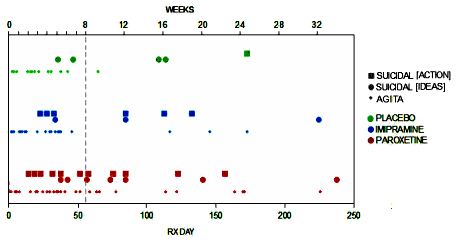
 Being asked to accept statements like that is particularly annoying from people who otherwise preach the gospel of evidence-based medicine from any available pulpit on any given Sunday. As a matter of fact, the whole point of doing RCTs in the first place is because people wouldn’t accept, "Newbery’s Brain Salt offers ‘A POSITIVE RELIEF AND CURE FOR Brain Troubles, Headaches, Sea Sickness,’ etc," particularly if the person saying it worked for F. Newbery & Sons.
Being asked to accept statements like that is particularly annoying from people who otherwise preach the gospel of evidence-based medicine from any available pulpit on any given Sunday. As a matter of fact, the whole point of doing RCTs in the first place is because people wouldn’t accept, "Newbery’s Brain Salt offers ‘A POSITIVE RELIEF AND CURE FOR Brain Troubles, Headaches, Sea Sickness,’ etc," particularly if the person saying it worked for F. Newbery & Sons.  revelations of widespread corruption in academic psychiatry [the KOLs] and volunteering in a clinic and being horrified at the medication regimens I found people on there. So I had a lot of catching up to do, luckily finding others who were willing to help. I think that the CROs [Contract Research Organizations], the Medical Writing Firms, that whole industry that entered the clinical trial scene must have been in its infancy about the time I was going into seclusion, because I didn’t know about any of it, though part of my reason for leaving had to do with a new administration that was keen on teaming up with PHARMA [another unfamiliar term]. I think of the time from going into practice until five or six years into my retirement as my "Rip Van Winkle" period:
revelations of widespread corruption in academic psychiatry [the KOLs] and volunteering in a clinic and being horrified at the medication regimens I found people on there. So I had a lot of catching up to do, luckily finding others who were willing to help. I think that the CROs [Contract Research Organizations], the Medical Writing Firms, that whole industry that entered the clinical trial scene must have been in its infancy about the time I was going into seclusion, because I didn’t know about any of it, though part of my reason for leaving had to do with a new administration that was keen on teaming up with PHARMA [another unfamiliar term]. I think of the time from going into practice until five or six years into my retirement as my "Rip Van Winkle" period: of meta-analyses.
of meta-analyses.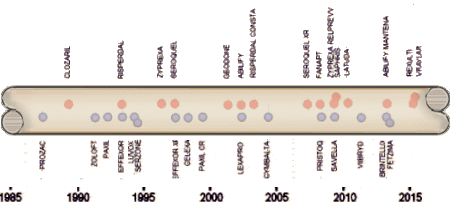





 While it’s discussed almost like it’s some complex governmental agency, clinicaltrials.gov is just a very large, online, searchable database of clinical trials using human subjects [see
While it’s discussed almost like it’s some complex governmental agency, clinicaltrials.gov is just a very large, online, searchable database of clinical trials using human subjects [see 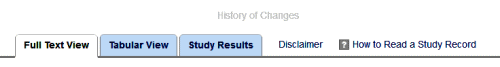
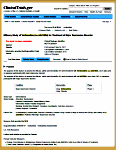 The opening screen [full text view] shows the information gathered when a trial is registered. It tells what’s being studied and why, who’s in charge of the trial, and who’s paying for it. It briefly defines the primary and secondary outcome parameters and how they’ll be analyzed at the end. There’s a section about eligibility and another identifying the location[s] of the study sites. There are lots of dates: when it was registered, when it was completed, etc [notably, it usually doesn’t say when the study actually began].
The opening screen [full text view] shows the information gathered when a trial is registered. It tells what’s being studied and why, who’s in charge of the trial, and who’s paying for it. It briefly defines the primary and secondary outcome parameters and how they’ll be analyzed at the end. There’s a section about eligibility and another identifying the location[s] of the study sites. There are lots of dates: when it was registered, when it was completed, etc [notably, it usually doesn’t say when the study actually began].
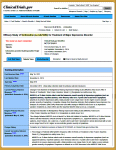 There’s another window into the same contents [tabular view] with a completely different layout. I’m usually visiting this site to see if they played by the rules, and so I find this view more helpful. For one thing, they report what the outcome variables were when the study was originally registered AND when the study was completed [for some reasons mentioned below, this isn’t always as helpful as it ought to be]. I think this view is the one most useful to scientists, physicians, and trialists – less "fishing around" required.
There’s another window into the same contents [tabular view] with a completely different layout. I’m usually visiting this site to see if they played by the rules, and so I find this view more helpful. For one thing, they report what the outcome variables were when the study was originally registered AND when the study was completed [for some reasons mentioned below, this isn’t always as helpful as it ought to be]. I think this view is the one most useful to scientists, physicians, and trialists – less "fishing around" required. 
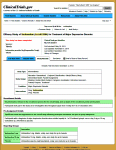 The results database has been the most disappointing aspect of this enterprise, not because of its structure, but because it has been essentially ignored – even in trials where it is legally mandated. Compliance percentages have been in the teens, even for NIMH/NIH funded trials, and close to zero for the contested studies I’ve tried to look at. The structural layout is fine – the results of the primary and secondary Outcomes and the Adverse Events. But it has way too often simply said
The results database has been the most disappointing aspect of this enterprise, not because of its structure, but because it has been essentially ignored – even in trials where it is legally mandated. Compliance percentages have been in the teens, even for NIMH/NIH funded trials, and close to zero for the contested studies I’ve tried to look at. The structural layout is fine – the results of the primary and secondary Outcomes and the Adverse Events. But it has way too often simply said 
 Clicking the link above the tabbed menu [History of Changes], you get a log of all the times it has been updated along the way, and a link that says "Continue to the history of changes for this study on the
Clicking the link above the tabbed menu [History of Changes], you get a log of all the times it has been updated along the way, and a link that says "Continue to the history of changes for this study on the  In the past, there have been reforms that should have worked, at least helped. They have failed us for several reasons, but one stands out. After the trumpets stopped blaring, they were just forgotten. The people doing the trials either didn’t do them, or didn’t do them right, or didn’t do them on time. The people in charge didn’t keep up with them or do anything in response to infractions if they even knew about them. They sounded good, but they flunked compliance, surveillance, and enforcement. So as we’re looking over these various changes and reforms, these provisions are paramount. Without these elements, it’s just another failed exercise.
In the past, there have been reforms that should have worked, at least helped. They have failed us for several reasons, but one stands out. After the trumpets stopped blaring, they were just forgotten. The people doing the trials either didn’t do them, or didn’t do them right, or didn’t do them on time. The people in charge didn’t keep up with them or do anything in response to infractions if they even knew about them. They sounded good, but they flunked compliance, surveillance, and enforcement. So as we’re looking over these various changes and reforms, these provisions are paramount. Without these elements, it’s just another failed exercise.