Posted on Wednesday 19 November 2014
The Chronicle of Higher EducationBy Paul BaskenNovember 19, 2014[behind pay-wall]
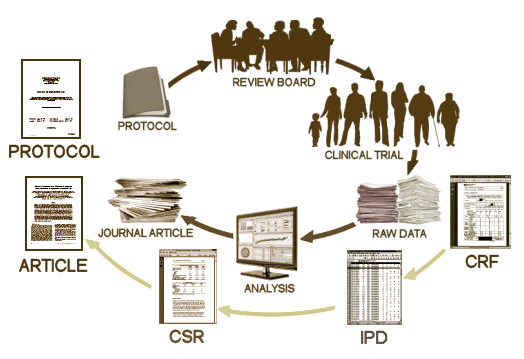
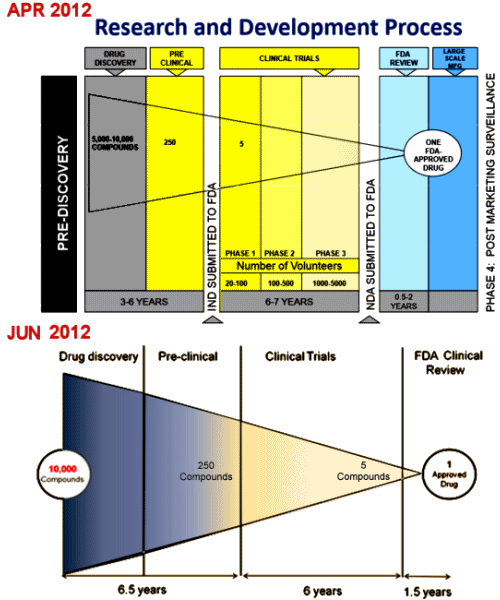
The Obama administration on Wednesday proposed new rules aimed at forcing researchers to publicly post details of medical trials involving human subjects, saying compliance to date has been woefully insufficient. For years, the government has required researchers using federal money, or seeking federal approval for a medical intervention, to announce the details of their trials in advance and post the results. But that hasn’t happened often enough, National Institutes of Health officials said. As evidence, they cited a study conducted at Yale University in 2012. Researchers studied a sample of trials using NIH money and found that fewer than half had results published within 30 months of the trials’ end.
"We as a community have a disappointing track record" of disseminating trial results, said Francis S. Collins, director of the NIH, in a briefing outlining the proposed new rules.Registering trials in advance and reporting the results afterward are critical to ensuring the integrity of studies, protecting patients, and building awareness of the results, Dr. Collins said. Under a pair of related proposals announced by NIH officials, researchers will be given specific guidelines on which trials are covered by the rules and which penalties will be levied against researchers who fail to comply. The current law is unspecific, NIH officials said. That has left researchers uncertain — or able to claim uncertainty — about the law’s exact requirements.Once the new rules take effect, "it will be a straightforward matter for us to be able to take compliance action," said Kathy L. Hudson, deputy director of science, outreach, and policy at NIH. The policy consists of two sets of complementary rules, one largely covering medical-trial results presented to the U.S. Food and Drug Administration as part of a drug-approval process, and the other covering research financed by the NIH.
In the case of the FDA, violations would lead to civil penalties. In the case of the NIH, researcher noncompliance could lead to the withholding of grant money, and loss of future agency support, Ms. Hudson said. "We’re certainly going to be taking that into account as we decide about future grant awards," she said of researcher-compliance records. The rules were issued with the expectation that they will take effect after a 90-day period of public comment…
National Institute of HealthNews ReleaseNovember. 19, 2014The U.S. Department of Health and Human Services today issued a Notice of Proposed Rulemaking [NPRM], which proposes regulations to implement reporting requirements for clinical trials that are subject to Title VIII of the Food and Drug Administration Amendments Act of 2007 [FDAAA]. The proposed rule clarifies requirements to clinical researchers for registering clinical trials and submitting summary trial results information to ClinicalTrials.gov, a publicly accessible database operated by the National Library of Medicine, part of the National Institutes of Health. A major proposed change from current requirements is the expansion of the scope of clinical trials required to submit summary results to include trials of unapproved, unlicensed, and uncleared products.
“Medical advances would not be possible without participants in clinical trials,” said NIH Director Francis S. Collins, M.D., Ph.D. “We owe it to every participant and the public at large to support the maximal use of this knowledge for the greatest benefit to human health. This important commitment from researchers to research participants must always be upheld.”
ClinicalTrials.gov currently contains registration information for more than 178,000 clinical trials and summary results for more than 15,000. These numbers include trials that are not subject to FDAAA. Among the primary benefits of registering and reporting results of clinical trials, including both positive and negative findings, is that it helps researchers prevent unnecessary duplication of trials, particularly when trial results indicate that a product under study may be unsafe or ineffective, and it establishes trust with clinical trial participants that the information from their participation is being put to maximum use to further knowledge about their condition.
Developed by NIH in close coordination with the FDA, the proposed rule details procedures for meeting the requirements established by FDAAA to improve public access to clinical trial information. FDAAA and the proposed rule apply to certain interventional studies of drugs, biological products, and devices that are regulated by the FDA, but, generally, not to phase 1 trials of drugs and biological products and small feasibility studies of devices. The proposed rule specifies how data collected and analyzed in a clinical trial would be required to be submitted to ClinicalTrials.gov. It would not affect requirements for the design or conduct of clinical trials or for the data that must be collected during clinical trials.
“This proposed rule would close an important gap, making additional information about clinical studies of investigational drugs, medical devices and biological products available to the public,” said FDA Commissioner Margaret A. Hamburg, M.D. “It would help eliminate unnecessary duplicative trials, advance biomedical innovation, and provide the public with a much richer understanding about the clinical trials for these products.”
Notable changes from current requirements and practice that are outlined in the proposed rule include:
JAMAby Kathy L. Hudson, PhD and Francis S. Collins, MD, MPHNovember 19, 2014
Pharmalot: WSJby Ed SilvermanNovember 19, 2014
Science: Science Insiderby By Jocelyn KaiserNovember 19, 2014


 I’m aware that an argument broke out in the comments that stepped outside the little box above the comments section – went interpersonal. It has happened before, which why that box says what it says. The first time, the circumstances were unusual. It was someone in town who discovered I had a blog and inserted himself into the comments as if it were a hot-line to my home. I shut down the blog [
I’m aware that an argument broke out in the comments that stepped outside the little box above the comments section – went interpersonal. It has happened before, which why that box says what it says. The first time, the circumstances were unusual. It was someone in town who discovered I had a blog and inserted himself into the comments as if it were a hot-line to my home. I shut down the blog [ The leader is no longer the leader, but survives and becomes just another vulnerable member. The safety of the group becomes the valued property and responsibility of the group itself. And safety without a shared enemy is the only viable principle for groups to remain healthy, creative, and endure. You saw the movie already. The Wizard of Oz. And that outcome is never guaranteed.
The leader is no longer the leader, but survives and becomes just another vulnerable member. The safety of the group becomes the valued property and responsibility of the group itself. And safety without a shared enemy is the only viable principle for groups to remain healthy, creative, and endure. You saw the movie already. The Wizard of Oz. And that outcome is never guaranteed. 
 When Sigmund Freud was in his 30’s, a young neurologist, he began to try to connect what he knew about the brain and what he saw in human behavior. The end of the 19th Century had seen a flowering of knowledge about the brain anatomy, micro-anatomy, and their correlation with the neurological syndromes. He wrote it as a treatise in 1895 he called
When Sigmund Freud was in his 30’s, a young neurologist, he began to try to connect what he knew about the brain and what he saw in human behavior. The end of the 19th Century had seen a flowering of knowledge about the brain anatomy, micro-anatomy, and their correlation with the neurological syndromes. He wrote it as a treatise in 1895 he called 
![Darrel Regier & David Kupfer [DSM-5], Allen Frances [DSM-4], John Feighner, Robert Spitzer [RDC, DSM-III, DSM-IIIR]](http://1boringoldman.com/images/thinkers-1.gif "Darrel Regier & David Kupfer [DSM-5], Allen Frances [DSM-4], John Feighner, Robert Spitzer [RDC, DSM-III, DSM-IIIR]")
![Tom Insel [Director NIMH] & Bruce Cuthbert [Director of Translational Research, NIMH]](http://1boringoldman.com/images/insel-cuthbert.gif "Tom Insel [Director NIMH] & Bruce Cuthbert [Director of Translational Research, NIMH]")


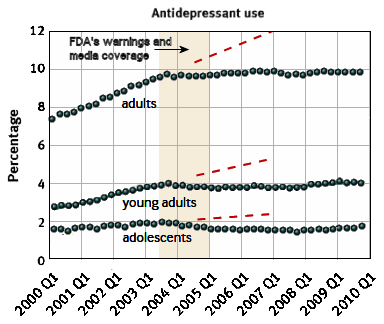
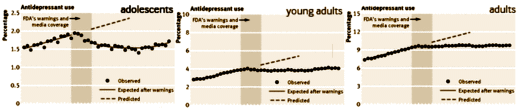
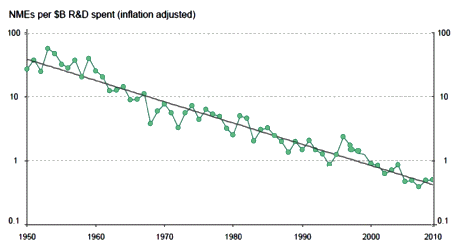
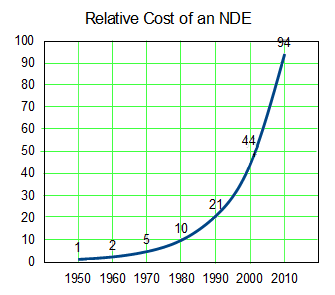

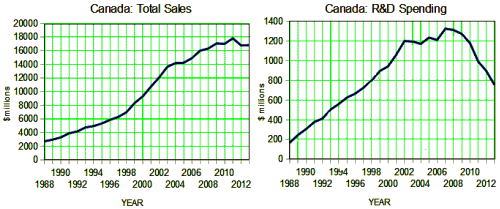
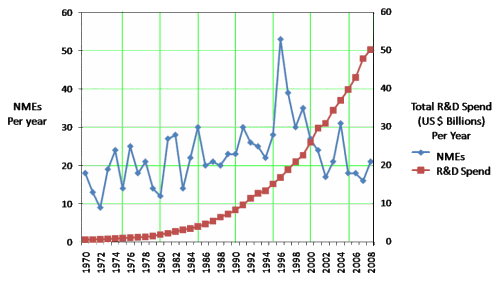
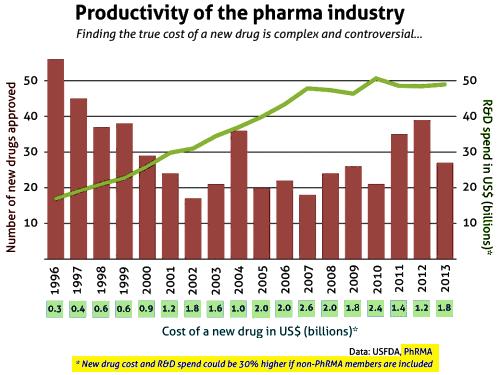
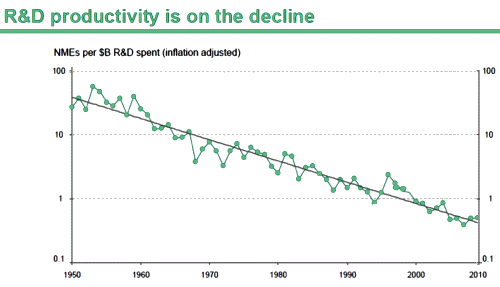
 The discovery of all three major classes of psychiatric drugs, antidepressants, antipsychotics, and anxiolytics, came about on the basis of serendipitous clinical observation. At the time of their discoveries, the mechanisms by which these molecules produce their effects were unknown, and it was only later that antipsychotics were shown to be D2 receptor antagonists, antidepressants monoamine reuptake inhibitors, and anxiolytics GABA receptor modulators. It is interesting and perhaps instructive to consider whether any of these classes of drugs could have been discovered by current drug discovery strategies. For example, what genetic or preclinical data exist that point to the D2 dopamine receptor as a likely target for antipsychotic activity? Presently there are no genetic data that suggest that this receptor is expressed or functions abnormally in psychotic disorders. And without the benefit of the prior clinical validation, it is difficult to see how preclinical data alone would point to the D2 receptor as an interesting potential target for the treatment of psychotic disorders. The same can be said for monoamine transporters with respect to depression where, like psychosis, there are no animal models based on disease pathophysiology and no compelling preclinical data pointing to these as potential targets for antidepressant drugs. This raises a troubling question: if in retrospect the three major classes of currently prescribed psychiatric drugs would likely never have been discovered using current drug discovery strategies, why should we believe that such strategies are likely to bear fruit now or in the future?…
The discovery of all three major classes of psychiatric drugs, antidepressants, antipsychotics, and anxiolytics, came about on the basis of serendipitous clinical observation. At the time of their discoveries, the mechanisms by which these molecules produce their effects were unknown, and it was only later that antipsychotics were shown to be D2 receptor antagonists, antidepressants monoamine reuptake inhibitors, and anxiolytics GABA receptor modulators. It is interesting and perhaps instructive to consider whether any of these classes of drugs could have been discovered by current drug discovery strategies. For example, what genetic or preclinical data exist that point to the D2 dopamine receptor as a likely target for antipsychotic activity? Presently there are no genetic data that suggest that this receptor is expressed or functions abnormally in psychotic disorders. And without the benefit of the prior clinical validation, it is difficult to see how preclinical data alone would point to the D2 receptor as an interesting potential target for the treatment of psychotic disorders. The same can be said for monoamine transporters with respect to depression where, like psychosis, there are no animal models based on disease pathophysiology and no compelling preclinical data pointing to these as potential targets for antidepressant drugs. This raises a troubling question: if in retrospect the three major classes of currently prescribed psychiatric drugs would likely never have been discovered using current drug discovery strategies, why should we believe that such strategies are likely to bear fruit now or in the future?…