I kind of liked writing the last post [about my connectomes…] and particularly the discussion that followed. I realize that over the recent years, I’ve written a lot about Clinical Trials, but my focus has been on the ways they’ve been misreported or distorted in the service of commerce. I’ve learned a lot about bias – for example Publication Bias [only publishing studies with the desired outcome]. I never realized the impact of leaving out negative studies. It’s analogous to omitting unwanted values in a single study – something you could never get away with. The more recent emphasis on meta-analyses has us looking at the family of studies as the data base rather than focusing on any single trial – highlighting the impact of Publication Bias.
 Randomized [placebo-controlled, double-blinded] Clinical Trials [RCTs] became a requirement for FDA Approval of new drugs in 1962 following the days of Thalidomide – adding proof of efficacy to the FDA’s original charge of insuring safety. I don’t really know the timeline of how FDA Approval moved from meaning not inert to being an actual endorsement – how we came to be reading things like "Prozac is now FDA Approved for the treatment of Major Depressive Disorder" in pharmaceutical ads [and in the Financial Times]. The FDA standard, not inert, is hardly a reasonable clinical treatment standard. It’s a mathematical or a chemical standard – separation from placebo certified by probability estimates. And in more cases than we’d like to admit, even those reported probabilities were improbable. Over time, this reform move aiming to curb corruption became its major super·highway.
Randomized [placebo-controlled, double-blinded] Clinical Trials [RCTs] became a requirement for FDA Approval of new drugs in 1962 following the days of Thalidomide – adding proof of efficacy to the FDA’s original charge of insuring safety. I don’t really know the timeline of how FDA Approval moved from meaning not inert to being an actual endorsement – how we came to be reading things like "Prozac is now FDA Approved for the treatment of Major Depressive Disorder" in pharmaceutical ads [and in the Financial Times]. The FDA standard, not inert, is hardly a reasonable clinical treatment standard. It’s a mathematical or a chemical standard – separation from placebo certified by probability estimates. And in more cases than we’d like to admit, even those reported probabilities were improbable. Over time, this reform move aiming to curb corruption became its major super·highway.
Randomized Placebo-Controlled, Double-Blinded Clinical Trials are intuitive. It makes perfect sense to look on a blinded comparison between drug versus no drug as the essence of evidence-based medicine. It’s hard to imagine an alternative. How did we even evaluate therapeutics prior to Lind’s comparison of citrus fruit to other treatments in Scurvy? One answer is that we went the other way. Instead of using the response of groups [as in a clinical trial] to inform the treatment of the individuals that come to our offices, we used what we learned from an individual to treat the groups that followed. My favorite disease is the one amateur carpenters get by hitting the thumb with a hammer. The blood under the nail causes excruciating pain, as does the drill kit in the ER used to release the pressure. Ever since an ER nurse showed me that burning a hole in the nail with an unfolded paper clip heated red hot was painless and got the job done, I’ve applied that treatment to every single subsequent case with stellar results. No Phase 3 Study required. It always works!
RCTs are really for evaluating safety, and then trying to sort out the real usefulness of treatments that work sometimes, or somewhat, or sometimes somewhat. I’ve been thinking the last couple of days that I might be winding up to go through some more of the math that people use to look at RTCs. It’s possible that something kind of amazing might be about to happen – data transparency. We’ve been thinking that if we had access to the actual data, we could make things like psycho·pharmacology into something right-sized. We’ve lived so long between a Scylla and a Charybdes level of black and white thinking, it’s interesting to ponder a world where the discourse is scientific rather than ideological, sectarian, financial, or just plain nasty.
A year ago, there was an article in the Lancet by Iain Chalmers, one of the founders of the Cochrame Collaboration, and Patrick Vallance, a President at GSK. It acknowledged helpful comments by Ben Goldacre in preparing the paper. They were talking about patient confidentiality in an era of data transparency, and suggested that putting "trialists" in the driver’s seat would be a solution because of their proven track record in protecting confidentiality:
by Patrick Vallance and Iain Chalmers
The Lancet 2013 382[9898]:1073 – 1074.
Publishing the results of all clinical trials, whoever funds them, is required for ethical, scientific, economic, and societal reasons. Individuals who take part in trials need to be sure that data they contribute are used to further knowledge, prevent unnecessary duplication of research, and improve the prospects for patients.
Endorsement of these principles is clear in the support received for the UK-based charitable trust Sense about Science’s campaign demanding that all clinical trials should be registered and reported. However, although the campaign recognises the advantages of analyses based on individual participant data (IPD), it is not calling for open access to IPD. The campaign recognises that risks to personal privacy must be taken seriously. These risks are not just theoretical: a recent study was able to identify 50 individuals from public websites that contained genetic information. The research community must work with others to define what constitutes appropriate protection of identifiable information if it is to retain public trust in the use of IPD.
Analyses based on IPD have many advantages. In 1970, The Lancet published a report based on nine trials of anticoagulant therapy after myocardial infarction. That study showed how, compared with analyses of aggregate data, access to IPD facilitated more thorough data checking; identified missing information; prompted renewed searches for key outcomes; enabled longitudinal analyses based on serial measurements in individuals; and offered greater reliability of subgroup analyses. Nearly two decades passed before others began to collaborate widely to use IPD analyses. These initiatives from collaborative trialists’ groups resulted in authoritative analyses of direct relevance to patient care in cancer and cardiovascular diseases, among others. The advantages of IPD analyses have prompted calls for wider access to such data, and we support these calls. However, robust arrangements are needed to minimise the risks of breaches of patient confidentiality. The experience gained within trialists’ collaborations is important, since, as far as we are aware, they have an unbroken record of maintaining patient confidentiality in their IPD analyses…
As much as I respect Drs. Chalmers and Goldacre, that article really pissed me off [
the wisdom of the Dixie Chicks…]. It felt like a Trojan Horse to me, a way to derail data transparency by making the data available to an elite cadre. And, by the way, I don’t agree that "patient confidentiality" applies to Clinical Trials. They’re subjects, not patients and I resent using an honored medical ethic to hide important parts of clinical research. But that’s what I thought then. What I think now is something a bit different that’s closer to the emotional reaction I had to that paper when it came out.
Having done a research fellowship in hard science, I knew more than the average psychiatrist about research methods and statistics, but I didn’t raise the questions I should’ve during the heyday of the SSRI/Atypical feeding fest of the 1990s and beyond. I had other things to study up on and I left the academic authors whose names were on those bylines to provide us with an accurate literature, or at least an honest literature. They didn’t do that [in spades]. It’s not that I suspect Dr. Chalmers and the other "trialists" of being like our now infamous psychiatric KOLs who made conflict of interest a way of life. It’s that I think it was and is my our responsibility to keep up with not just the literature, but to have at least an ongoing working understanding of the scientific methodology driving it. No more delegating to the scientific elite. There were three prominent Department Chairmen on Senator Grassley’s COI list, one of whom was an APA president elect. It’s as simple as the saying, "Fool me once, shame on you. Fool me twice, shame on me."
Writing this blog has lead me to "bone up" on my statistics, and I feel comfortable with that part. But what Dr. Carroll called clinimetrics has been new territory for me and I’m self-taught with help where I can find it. I enjoyed writing about a piece of it last time – effect sizes. It was helpful to put what I thought down on paper, and really helpful to read the comments from people with a more experiential acquaintance or just an interest. I think I’ll do some more of that. Even if I get some of it wrong, it’ll help to have others get my wheels more on the tracks.
If the long sought data transparency finally does come our way, we need to know what to do with it. It’s important going forward, and equally important looking backwards. There’s a lot of flotsam and jetsam still floating around out there from the previous shipwrecks that has to do with drugs still being used by the buckets·full now they’re in the generic domain and affordable. Ben Goldacre famously said that "the best disinfectant is sunlight." But that’s only true if a lot of us know what we’re looking at…




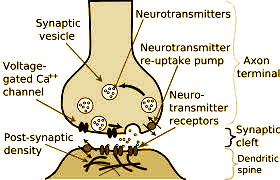 [like poisons or blood pressure pills]. If we talked about neurotransmitters [or much of anything else] in the grey-matter, I don’t recall it. That chatter came ten years later in psychiatry, where no drug lecture was complete without what I call the "NIH memorial nerve ending" slide, recycling neurotransmitters of various ilks [later, I had such a slide of my very own].
[like poisons or blood pressure pills]. If we talked about neurotransmitters [or much of anything else] in the grey-matter, I don’t recall it. That chatter came ten years later in psychiatry, where no drug lecture was complete without what I call the "NIH memorial nerve ending" slide, recycling neurotransmitters of various ilks [later, I had such a slide of my very own]. At this point, I could go off on a diatribe about the difference between the Dogmatists and the Skeptics in ancient Greece. In fact, maybe I’ll do just do that. I suppose if you have a favorite Greek philosopher, you can talk about him more than once. So I’ll borrow from a short former
At this point, I could go off on a diatribe about the difference between the Dogmatists and the Skeptics in ancient Greece. In fact, maybe I’ll do just do that. I suppose if you have a favorite Greek philosopher, you can talk about him more than once. So I’ll borrow from a short former  Pyrrho [Πuρρων, c. 360 BC – c. 270 BC]:
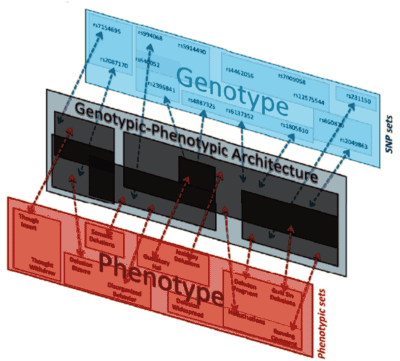
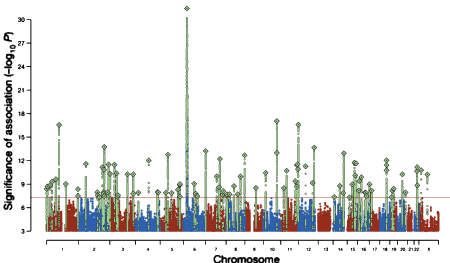
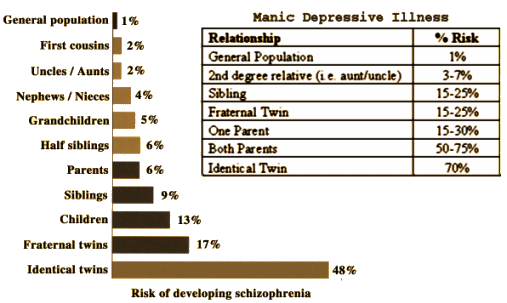
Pyrrho [Πuρρων, c. 360 BC – c. 270 BC]: As much as I like to talk about Data Transparency and try to demystify some of the calculations used in the Clinical Trials, when it comes to the modern genetic studies, I’m lost at sea. They’re doing calculus and I’m still on the multiplication tables just over counting on my fingers. I know that GWAS stands for genome-wide association study, and I sort of know what a SNP [single-nucleotide polymorphism] is. Beyond that, I’m only dimly able to understand how a given study was conducted and what the authors make of their findings, but not able to speak to the quality of the work. So in this case, I’ll say what a SNP is, because these geneticists can talk of little else, and if you talk genetics, you have to throw in SNP [pronounced "snip"] every so often to qualify yourself. Remember that the genome [the sequence of nucleotides on the chromosomes] fills a pretty large cabinet and makes for some really monotonous reading.
As much as I like to talk about Data Transparency and try to demystify some of the calculations used in the Clinical Trials, when it comes to the modern genetic studies, I’m lost at sea. They’re doing calculus and I’m still on the multiplication tables just over counting on my fingers. I know that GWAS stands for genome-wide association study, and I sort of know what a SNP [single-nucleotide polymorphism] is. Beyond that, I’m only dimly able to understand how a given study was conducted and what the authors make of their findings, but not able to speak to the quality of the work. So in this case, I’ll say what a SNP is, because these geneticists can talk of little else, and if you talk genetics, you have to throw in SNP [pronounced "snip"] every so often to qualify yourself. Remember that the genome [the sequence of nucleotides on the chromosomes] fills a pretty large cabinet and makes for some really monotonous reading.
 I like Dr. Summergrad. He doesn’t lead with the arrogance of his recent predecessors, doesn’t make assumptions about the primacy of psychiatry in the mental health cosmology, and while he wears a white coat – he wears it loosely. But it will take more than a long absent humility to achieve "explicit recognition of the psychiatric treatment needs for Americans suffering from mental illness and substance use disorders, and in particular for the 13 million Americans who suffer from debilitating serious mental illnesses" primarily because for thirty years, psychiatry has itself had a monocular view of those needs. He speaks against SAMHSA coming down on one side of "the divide," yet psychiatry itself has helped to create and both actively and passively nurtured the division.
I like Dr. Summergrad. He doesn’t lead with the arrogance of his recent predecessors, doesn’t make assumptions about the primacy of psychiatry in the mental health cosmology, and while he wears a white coat – he wears it loosely. But it will take more than a long absent humility to achieve "explicit recognition of the psychiatric treatment needs for Americans suffering from mental illness and substance use disorders, and in particular for the 13 million Americans who suffer from debilitating serious mental illnesses" primarily because for thirty years, psychiatry has itself had a monocular view of those needs. He speaks against SAMHSA coming down on one side of "the divide," yet psychiatry itself has helped to create and both actively and passively nurtured the division.
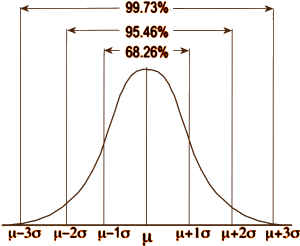 Over there on the right is our old friend, Mr. Normal Distribution. If you take a large sample of almost anything, the values will look like this with the measurement variable across the bottom [abscissa] and the frequency of the values on the left axis [ordinate]. So you can describe a dataset that fits this distribution [most of them] with just three numbers: μ [the mean, average]; n [the number of observations]; and σ [the standard deviation, an index of how much variability there is within the data]. 95% of the values fall between two standard deviations on either side of the mean. That means that measurements outside those limits have only a 5% chance of belonging to this group – thus p < 0.05 [p as in probability].
Over there on the right is our old friend, Mr. Normal Distribution. If you take a large sample of almost anything, the values will look like this with the measurement variable across the bottom [abscissa] and the frequency of the values on the left axis [ordinate]. So you can describe a dataset that fits this distribution [most of them] with just three numbers: μ [the mean, average]; n [the number of observations]; and σ [the standard deviation, an index of how much variability there is within the data]. 95% of the values fall between two standard deviations on either side of the mean. That means that measurements outside those limits have only a 5% chance of belonging to this group – thus p < 0.05 [p as in probability]. 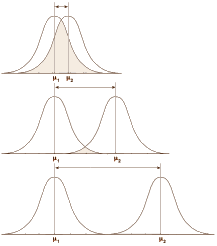 In the examples on the left, all three might be statistically significant but looking at the p value won’t tell you anything about the likelihood it will be a property noticed by individual patients. It might be simple a chemical finding that’s imperceptible, or it might be a power-house. But p doesn’t tell you that. Regulatory bodies, like the Food and Drug Administration [FDA], were established to keep inactive patent medicines off the medical market, not to direct drug usage or attest to the magnitude of their effects. So the FDA insists on two well conducted Clinical Trials that demonstrate statistically significant differences between the drug and a placebo. The main task of the FDA Approval process is safety – what are the drug’s potential harms? Efficacy is a soft standard added on in 1962.
In the examples on the left, all three might be statistically significant but looking at the p value won’t tell you anything about the likelihood it will be a property noticed by individual patients. It might be simple a chemical finding that’s imperceptible, or it might be a power-house. But p doesn’t tell you that. Regulatory bodies, like the Food and Drug Administration [FDA], were established to keep inactive patent medicines off the medical market, not to direct drug usage or attest to the magnitude of their effects. So the FDA insists on two well conducted Clinical Trials that demonstrate statistically significant differences between the drug and a placebo. The main task of the FDA Approval process is safety – what are the drug’s potential harms? Efficacy is a soft standard added on in 1962.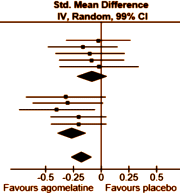 There are no absolute standards for a meaningful Standardized Mean Difference. Cohen suggested 0.25 = weak; 0.50 = moderate; 0.75 = strong. But you could gather a bunch of statisticians and they could argue about that for the whole evening. The place where Effect Sizes are routinely used is in meta-analyses that compared multiple studies and/or multiple drugs. I guess you could say it’s a powerful relativity tool, often combined with a representation of the 95% Confidence Interval. In the example on the right from the article quoted above, the Standardized Mean Difference is on the Abscissa with the Confidence Intervals as the horizontal line [in this example, the 99% Confidence Intervals were used]. This is the format used in the Cochrane Systematic Reviews called a forest plot and it tells us a lot. The top 5 studies are unpublished Clinical Trials of agomelatine vs placebo. The weighted average is 0.08 in favor of agomelatine [which might as well be zero], and none are significant at the 1% level [the Confidence Interval line crosses zero]. The bottom 5 studies are published studies of agomelatine. The weighted average is 0.26 with only one significant at the 1% level. Overall, the weighted average Standardized Mean Difference is 0.18. In everyday language, a trivial effect with clear publication bias.
There are no absolute standards for a meaningful Standardized Mean Difference. Cohen suggested 0.25 = weak; 0.50 = moderate; 0.75 = strong. But you could gather a bunch of statisticians and they could argue about that for the whole evening. The place where Effect Sizes are routinely used is in meta-analyses that compared multiple studies and/or multiple drugs. I guess you could say it’s a powerful relativity tool, often combined with a representation of the 95% Confidence Interval. In the example on the right from the article quoted above, the Standardized Mean Difference is on the Abscissa with the Confidence Intervals as the horizontal line [in this example, the 99% Confidence Intervals were used]. This is the format used in the Cochrane Systematic Reviews called a forest plot and it tells us a lot. The top 5 studies are unpublished Clinical Trials of agomelatine vs placebo. The weighted average is 0.08 in favor of agomelatine [which might as well be zero], and none are significant at the 1% level [the Confidence Interval line crosses zero]. The bottom 5 studies are published studies of agomelatine. The weighted average is 0.26 with only one significant at the 1% level. Overall, the weighted average Standardized Mean Difference is 0.18. In everyday language, a trivial effect with clear publication bias.