This article [Schwartz et al] compares the results found in the ClinicalTrials.gov Results Database with those in Drugs@FDA for 100 drugs up for approval between January 2013 and June 2014. It makes little sense to look at it outside the context of a whole series of articles focusing on ClinicalTrials.gov going back in time. At the end of this post, there are seven abstracts of a representative group of such articles, arranged by year of publication [all available full text on-line]. You might just scan the conclusions before you start…
by Lisa Schwartz, Steven Woloshin, Eugene Zheng, Tony Tse, and Deborah Zarin
Annals of Internal Medicine. 2016 165:421-430.
Background: Pharmaceutical companies and other trial sponsors must submit certain trial results to ClinicalTrials.gov. The validity of these results is unclear.
Purpose: To validate results posted on ClinicalTrials.gov against publicly available U.S. Food and Drug Administration [FDA] reviews on Drugs@FDA.
Data Sources: ClinicalTrials.gov [registry and results database] and Drugs@FDA [medical and statistical reviews].
Study Selection: 100 parallel-group, randomized trials for new drug approvals [January 2013 to July 2014] with results posted on ClinicalTrials.gov [15 March 2015].
Data Extraction: 2 assessors extracted, and another verified, the trial design, primary and secondary outcomes, adverse events, and deaths.
Results: Most trials were phase 3 [90%], double-blind [92%], and placebo-controlled [73%] and involved 32 drugs from 24 companies. Of 137 primary outcomes identified from ClinicalTrials.gov, 134 [98%] had corresponding data at Drugs@FDA, 130 [95%] had concordant definitions, and 107 [78%] had concordant results. Most differences were nominal [that is, relative difference <10%]. Primary outcome results in 14 trials could not be validated. Of 1927 secondary outcomes from ClinicalTrials.gov, Drugs@FDA mentioned 1061 [55%] and included results data for 367 [19%]. Of 96 trials with 1 or more serious adverse events in either source, 14 could be compared and 7 had discordant numbers of persons experiencing the adverse events. Of 62 trials with 1 or more deaths in either source, 25 could be compared and 17 were discordant.
Limitation: Unknown generalizability to uncontrolled or cross-over trial results.
Conclusion: Primary outcome definitions and results were largely concordant between ClinicalTrials.gov and Drugs@FDA. Half the secondary outcomes, as well as serious events and deaths, could not be validated because Drugs@FDA includes only "key outcomes" for regulatory decision making and frequently includes only adverse event results aggregated across multiple trials.
In response to the AIDS activists’ insistence on rapid access to drug trials, in 1988 Congress mandated the creation of the
AIDS Clinical Trials Information System [
ACTIS]. It was a good idea and over time lead to the creation of
ClinicalTrials.gov, an online database maintained by the
NIH National Library of Medicine. By 2007 [
Food and Drug Administration Amendments Act of 2007], both
Registration and
Results reporting on
ClinicalTrials.gov became mandatory for all FDA regulated drugs.
But they just didn’t do it. Most trials were registered but often long after they started, and the ClinicalTrials.gov Results Database was the loneliest database on the Internet. There was no enforcement, even from Journal Editors who also claimed to require it. I’ve collected a few of the REFERENCES below that document the problems from several different angles. But even done right [Registered a priori with Results submitted], there are still difficulties. So this system created to promote public transparency just hasn’t worked, and the consequences have become the stuff of legends. Recently, the NIH, ClinicalTrials.gov, and the FDA rolled out a set of reforms designed to correct the things in the ClinicalTrials.gov system. While the effort is appreciated, it was primarily focused on compliance and enforcement – more a commitment to actually do what was supposed to happen in 2007, but uninformed by the multiple real problems in the system today. For example, nobody checks the submitted information, and the deadlines for submission mean that, at best, the information comes after it’s actually needed. On-time is too-late.
So we have a proposal to Congress known as
The Petition. It has several parts:
-
At the time a clinical trial is Registered on ClinicalTrials.gov, the Primary and Secondary Outcome Parameters and the Methods by which they will be analyzed need to be included – no lag time. We ask that the FDA certify that they are concordant with the Protocol and Statistical Analysis Plan which they usually have as part of the FDA Registration. The "why?" is obvious. The most common method of spinning the results is to change the outcome variables. Requiring them in black and white at the time of Registration anchors them so any attempt to change them will be obvious. And "why the FDA?" It is the agency responsible to us to insure the integrity of scientific basis of our pharmacopoeia. It’s their job.
-
After the study is completed, the ClinicalTrials.gov Results Database must be completed at the time of any submission to the FDA with the results of the prespecified outcomes and analyses clearly labeled. Other outcomes listed should be identified as "non-protocol" or "exploratory." Again, the FDA needs to certify that the ClinicalTrials.gov results are concordant with those submitted to the FDA. No lag time. If they have results to submit to the FDA, they have them to submit them to ClinicalTrials.gov as well.
-
While the FDA/NIH has no place in telling Journals what to publish, any publication or advertisement that results from a trial and uses non-protocol outcomes should make that clear, otherwise it will be considered false advertisement of an FDA regulated drug. Academic Journal articles of clinical trials are advertisements
-
If the FDA recommends further trials as part of an approval, it is the responsibility of the FDA to insure that these trials are done expeditiously and follow the same procedures as the original NDA.
Congress instantiated ClinicalTrials.gov in 1997 trying to add a public interface to the clinical trial system. In 2007, Congress mandated that ClinicalTrial.gov become the required public interface to our clinical trial system but that mandate was ignored until the complaints were so loud as to be heard around the world. It’s time for Congress to finally close the deal in 2017. One might look at the paper above [Schwartz et al] as a sign that compliance in recent years has improved. A much more realistic interpretation would be that they are now being so closely watched that they’ve finally begun to comply. We absolutely must have that kind of oversight built-in to this system to insure that the rampant corruption we’ve had in plain sight comes to an immediate halt. Look at the sample articles in the REFERENCES to see what happens when no one is looking.
There is no reason for any lag time in posting the needed information at the time of Registration or Submission. They have it at hand, and we need it. Editors, Peer Reviewers, readers, everyone has the right to see the results certified by the responsible party. That responsible party is the FDA. They are part of the problem in colluding with industry to keep the raw data secret. Perhaps they have no choice. But that sure doesn’t apply to the a priori Protocol or the Protocol-directed Results. So sign the The Petition now and send it to everyone you can think of. It’s time for Congress to act!
REFERENCES
2009
by Mathieu S, Boutron I, Moher D, Altman DG, and Ravaud P.
JAMA. 2009 302[9]:977-984.
Context: As of 2005, the International Committee of Medical Journal Editors required investigators to register their trials prior to participant enrollment as a precondition for publishing the trial’s findings in member journals.
Objective: To assess the proportion of registered trials with results recently published in journals with high impact factors; to compare the primary otcomes specified in trial registries with those reported in the published articles; and to determine whether primary outcome reporting bias favored significant outcomes.
Data Sources: MEDLINE via PubMed was searched for reports of randomized controlled trials [RCTs] in 3 medical areas [cardiology, rheumatology, and gastroenterology] indexed in 2008 in the 10 general medical journals and specialty journals with the highest impact factors.
Data Extraction: For each included article, we obtained the trial registration information using a standardized data extraction form.
Results: Of the 323 included trials, 147 [45.5%] were adequately registered [ie, registered before the end of the trial, with the primary outcome clearly specified]. Trial registration was lacking for 89 published reports [27.6%], 45 trials [13.9%] were registered after the completion of the study, 39 [12%] were registered with no or an unclear description of the primary outcome, and 3 [0.9%] were registered after the completion of the study and had an unclear description of the primary outcome. Among articles with trials adequately registered, 31% [46 of 147] showed some evidence of discrepancies between the outcomes registered and the outcomes published. The influence of these discrepancies could be assessed in only half of them and in these statistically significant results were favored in 82.6% [19 of 23].
Conclusion: Comparison of the primary outcomes of RCTs registered with their subsequent publication indicated that selective outcome reporting is prevalent.
by Joseph S. Ross, Gregory K. Mulvey, Elizabeth M. Hines, Steven E. Nissen, and Harlan M. Krumholz
PLoS Medicine. 2009 6[9]: e1000144.
Background: ClinicalTrials.gov is a publicly accessible, Internet-based registry of clinical trials managed by the US National Library of Medicine that has the potential to address selective trial publication. Our objectives were to examine completeness of registration within ClinicalTrials.gov and to determine the extent and correlates of selective publication
Methods and Findings: We examined reporting of registration information among a cross-section of trials that had been registered at ClinicalTrials.gov after December 31, 1999 and updated as having been completed by June 8, 2007, excluding phase I trials. We then determined publication status among a random 10% subsample by searching MEDLINE using a systematic protocol, after excluding trials completed after December 31, 2005 to allow at least 2 y for publication following completion. Among the full sample of completed trials [n=7,515], nearly 100% reported all data elements mandated by ClinicalTrials.gov, such as intervention and sponsorship. Optional data element reporting varied, with 53% reporting trial end date, 66% reporting primary outcome, and 87% reporting trial start date. Among the 10% subsample, less than half [311 of 677, 46%] of trials were published, among which 96 [31%] provided a citation within ClinicalTrials.gov of a publication describing trial results. Trials primarily sponsored by industry [40%, 144 of 357] were less likely to be published when compared with nonindustry/nongovernment sponsored trials [56%, 110 of 198; p<0.001], but there was no significant difference when compared with government sponsored trials [47%, 57 of 122; p=0.22]. Among trials that reported an end date, 75 of 123 [61%] completed prior to 2004, 50 of 96 [52%] completed during 2004, and 62 of 149 [42%] completed during 2005 were published [p=0.006].
Conclusions: Reporting of optional data elements varied and publication rates among completed trials registered within ClinicalTrials.gov were low. Without greater attention to reporting of all data elements, the potential for ClinicalTrials.gov to address selective publication of clinical trials will be limited.
2011
by Michael R. Law, Yuko Kawasumi, and Steven G. Morgan
Health Affairs. 2011 30[12]:2338-2345.
Clinical trial registries are public databases created to prospectively document the methods and measures of prescription drug studies and retrospectively collect a summary of results. In 2007 the US government began requiring that researchers register certain studies and report the results on ClinicalTrials.gov, a public database of federally and privately supported trials conducted in the United States and abroad. We found that although the mandate briefly increased trial registrations, 39 percent of trials were still registered late after the mandate’s deadline, and only 12 percent of completed studies reported results within a year, as required by the mandate. This result is important because there is evidence of selective reporting even among registered trials. Furthermore, we found that trials funded by industry were more than three times as likely to report results than were trials funded by the National Institutes of Health. Thus, additional enforcement may be required to ensure disclosure of all trial results, leading to a better understanding of drug safety and efficacy. Congress should also reconsider the three-year delay in reporting results for products that have been approved by the Food and Drug Administration and are in use by patients.
by Deborah A. Zarin, Tony Tse, Rebecca J. Williams, Robert M. Califf, and Nicholas C. Ide
New England Journal of Medicine. 2011 364:852-860.
Background: The ClinicalTrials.gov trial registry was expanded in 2008 to include a database for reporting summary results. We summarize the structure and contents of the results database, provide an update of relevant policies, and show how the data can be used to gain insight into the state of clinical research.
Methods: We analyzed ClinicalTrials.gov data that were publicly available between September 2009 and September 2010.
Results: As of September 27, 2010, ClinicalTrials.gov received approximately 330 new and 2000 revised registrations each week, along with 30 new and 80 revised results submissions. We characterized the 79,413 registry and 2178 results of trial records available as of September 2010. From a sample cohort of results records, 78 of 150 [52%] had associated publications within 2 years after posting. Of results records available publicly, 20% reported more than two primary outcome measures and 5% reported more than five. Of a sample of 100 registry record outcome measures, 61% lacked specificity in describing the metric used in the planned analysis. In a sample of 700 results records, the mean number of different analysis populations per study group was 2.5 [median, 1; range, 1 to 25]. Of these trials, 24% reported results for 90% or less of their participants.
Conclusions: ClinicalTrials.gov provides access to study results not otherwise available to the public. Although the database allows examination of various aspects of ongoing and completed clinical trials, its ultimate usefulness depends on the research community to submit accurate, informative data.
2012
by Christopher J Gill
BMJ Open. 2012 2:e001186
Context: The Food and Drug Administration Modernization Act of 1997 [FDAMA] and the FDA Amendment Act of 2007 [FDAAA], respectively, established mandates for registration of interventional human research studies on the website clinicaltrials.gov [CTG] and for posting of results of completed studies.
Objective: To characterise, contrast and explain rates of compliance with ontime registration of new studies and posting of results for completed studies on CTG.
Design: Statistical analysis of publically available data downloaded from the CTG website.
Participants: US studies registered on CTG since 1 November 1999, the date when the CTG website became operational, through 24 June 2011, the date the data set was downloaded for analysis.
Main outcome measures: Ontime registration [within 21 days of study start]; average delay from study start to registration; proportion of studies posting their results from within the group of studies listed as completed on CTG.
Results: As of 24 June 2011, CTG contained 54?890 studies registered in the USA. Prior to 2005, an estimated 80% of US studies were not being registered. Among registered studies, only 55.7% registered within the 21-day reporting window. The average delay on CTG was 322 days. Between 28 September 2007 and June 23 2010, 28% of intervention studies at Phase II or beyond posted their study results on CTG, compared with 8.4% for studies without industry funding [RR 4.2, 95% CI 3.7 to 4.8]. Factors associated with posting of results included exclusively paediatric studies [adjusted OR [AOR] 2.9, 95% CI 2.1 to 4.0], and later phase clinical trials [relative to Phase II studies, AOR for Phase III was 3.4, 95% CI 2.8 to 4.1; AOR for Phase IV was 6.0, 95% CI 4.8 to 7.6].
Conclusions: Non-compliance with FDAMA and FDAAA appears to be very common, although compliance is higher for studies sponsored by industry. Further oversight may be required to improve compliance.
2013
by Jones CW, Handler L, Crowell KE, Keil LG, Weaver MA, and Platts-Mills TF.
British Medical Journal. 2013 347:f6104.
Objective: To estimate the frequency with which results of large randomized clinical trials registered with ClinicalTrials.gov are not available to the public.
Design: Cross sectional analysis
Setting: Trials with at least 500 participants that were prospectively registered with ClinicalTrials.gov and completed prior to January 2009.
Data Sources: PubMed, Google Scholar, and Embase were searched to identify published manuscripts containing trial results. The final literature search occurred in November 2012. Registry entries for unpublished trials were reviewed to determine whether results for these studies were available in the ClinicalTrials.gov results database.
Main Outcome Sources: The frequency of non-publication of trial results and, among unpublished studies, the frequency with which results are unavailable in the ClinicalTrials.gov database.
Results: Of 585 registered trials, 171 [29%] remained unpublished. These 171 unpublished trials had an estimated total enrollment of 299,763 study participants. The median time between study completion and the final literature search was 60 months for unpublished trials. Non-publication was more common among trials that received industry funding [150/468, 32%] than those that did not [21/117, 18%], P=0.003. Of the 171 unpublished trials, 133 [78%] had no results available in ClinicalTrials.gov.
Conclusions: Among this group of large clinical trials, non-publication of results was common and the availability of results in the ClinicalTrials.gov database was limited. A substantial number of study participants were exposed to the risks of trial participation without the societal benefits that accompany the dissemination of trial results.
2014
by Jessica E. Becker, Harlan M. Krumholz, Gal Ben-Josef, and Joseph S. Ross
JAMA. 2014 311[10]:1063-1065.
The 2007 Food and Drug Administration [FDA] Amendments Act expanded requirements for ClinicalTrials.gov, a public clinical trial registry maintained by the National Library of Medicine, mandating results reporting within 12 months of trial completion for all FDA-regulated medical products. Reporting of mandatory trial registration information on ClinicalTrials.gov is fairly complete, although there are concerns about its specificity; optional trial registration information is less complete.1- 4 To our knowledge, no studies have examined reporting and accuracy of trial results information. Accordingly, we compared trial information and results reported on ClinicalTrials.gov with corresponding peer-reviewed publications.
Methods: We conducted a cross-sectional analysis of clinical trials for which the primary results were published between July 1, 2010, and June 30, 2011, in Medline-indexed, high-impact journals [impact factor ≥10; Web of Knowledge, Thomson Reuters] and that were registered on ClinicalTrials.gov and reported results. For each trial, we assessed reporting of the following results information on ClinicalTrials.gov and corresponding publications and compared reported information in both sources: cohort characteristics [enrollment and completion, age/sex demographics], trial intervention, and primary and secondary efficacy end points and results. Results information was considered concordant if the described end point, time of ascertainment, and measurement scale matched. Reported results were categorized as concordant [ie, numerically equal], discordant [ie, not numerically equal], or could not be compared [ie, reported numerically in one, graphically in the other]. For discordant primary efficacy end points, we determined whether the discrepancy altered study interpretation. Descriptive analyses were performed using Excel [version 14.3.1, Microsoft].
Results: We identified 96 trials reporting results on ClinicalTrials.gov that were published in 19 high-impact journals. For 70 trials [73%], industry was the lead funder. The most common conditions studied were cardiovascular disease, diabetes, and hyperlipidemia [n = 21; 23%]; cancer [n = 20; 21%]; and infectious disease [n = 19; 20%]. Trials were most frequently published by New England Journal of Medicine [n = 23; 24%], Lancet [n = 18; 19%], and JAMA [n = 11; 12%]. Cohort, intervention, and efficacy end point information was reported for 93% to 100% of trials in both sources [Table 1]. However, 93 of 96 trials had at least 1 discordance among reported trial information or reported results.
Among trials reporting each cohort characteristic and trial intervention information, discordance ranged from 2% to 22% and was highest for completion rate and trial intervention, for which different descriptions of dosages, frequencies, or duration of intervention were common.
There were 91 trials defining 156 primary efficacy end points [5 trials defined only primary safety end points], 132 [85%] of which were described in both sources, 14 [9%] only on ClinicalTrials.gov, and 10 [6%] only in publications. Among 132 end points described in both sources, results for 30 [23%] could not be compared and 21 [16%] were discordant. The majority [n = 15] of discordant results did not alter trial interpretation, although for 6, the discordance did [Table 2]. Overall, 81 of 156 [52%] primary efficacy end points were described in both sources and reported concordant results.
There were 96 trials defining 2089 secondary efficacy end points, 619 [30%] of which were described in both sources, 421 [20%] only on ClinicalTrials.gov, and 1049 [50%] only in publications. Among 619 end points described in both sources, results for 228 [37%] could not be compared, whereas 53 [9%] were discordant. Overall, 338 of 2089 [16%] secondary efficacy end points were described in both sources and reported concordant results.
Discussion: Among clinical trials published in high-impact journals that reported results on ClinicalTrials.gov, nearly all had at least 1 discrepancy in the cohort, intervention, or results reported between the 2 sources, including many discordances in reported primary end points. For discordances observed when both the publication and ClinicalTrials.gov reported the same end point, possible explanations include reporting and typographical errors as well as changes made during the course of the peer review process. For discordances observed when one source reported a result but not the other, possible explanations include journal space limitations and intentional dissemination of more favorable end points and results in publications.
Our study was limited to a small number of trials that were not only registered and reported results, but also published in high-impact journals. However, because articles published in high-impact journals are generally the highest-quality research studies and undergo more rigorous peer review, the trials in our sample likely represent best-case scenarios with respect to the quality of results reporting. Our findings raise questions about accuracy of both ClinicalTrials.gov and publications, as each source’s reported results at times disagreed with the other. Further efforts are needed to ensure accuracy of public clinical trial result reporting efforts.


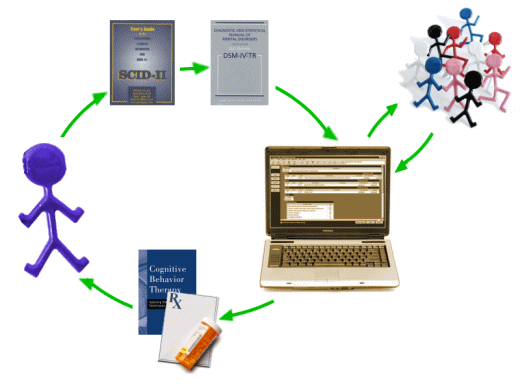 Back then, I drew a little diagram to illustrate what I was reading. A structured interview [SCID] lead to a diagnosis [DSM-IV]. Enter that into Dr. Madhukar Trivedi‘s computer program and it spit out an evidence-based treatment informed by the latest RTCs [Randomized Clinical Trials]. The evidence-based treatments for depression provided were either the second generation antidepressants or CBT [Cognitive Behavior Therapy]. And then you went round and round the process hopefully iterating towards success. The actual article I was reading had Dr. Trivedi‘s reflections about why the clinicians ignored his system unless he was actively looking over their shoulder. I had no difficulty at all understanding that myself [but I’ll fight the temptation to say why yet again]. But that was then, and this is now.
Back then, I drew a little diagram to illustrate what I was reading. A structured interview [SCID] lead to a diagnosis [DSM-IV]. Enter that into Dr. Madhukar Trivedi‘s computer program and it spit out an evidence-based treatment informed by the latest RTCs [Randomized Clinical Trials]. The evidence-based treatments for depression provided were either the second generation antidepressants or CBT [Cognitive Behavior Therapy]. And then you went round and round the process hopefully iterating towards success. The actual article I was reading had Dr. Trivedi‘s reflections about why the clinicians ignored his system unless he was actively looking over their shoulder. I had no difficulty at all understanding that myself [but I’ll fight the temptation to say why yet again]. But that was then, and this is now. Something else I encountered for the first time on awakening – the
Something else I encountered for the first time on awakening – the