Posted on Friday 1 January 2016



I am personally drawn to the mathematical/statistical analyses employed in a properly conducted Randomized Double Blind Clinical Trial [RCT], and would like to think that if we had access to the raw data from Clinical Trials [Data Transparency], we could generate an accurate efficacy and safety profile for our drugs based that data. That was the general gist of the first ending to this story. In that scenario, the carefully analyzed RCT would become the gold standard for evaluating medications. Certainly, the RCT is, by Congressional Decree, already the gold standard for the FDA’s Approval process.
There are other ways to look at this [see hearing voices… and something essential betrayed…]. While the RCT may be a reasonable way for the FDA to evaluate new drugs for approval, that doesn’t necessarily translate to a standard for clinical practice [see this comment by Dr. Bernard Carroll and Pharmageddon by Dr. David Healy]. Subjects are screened for exclusions. Subjects are recruited and often paid whereas Patients are help-seeking. The RCTs are short-term [6-8 weeks] while Patients are on medications much longer [these days, sometimes indefinitely]. Subjects are seen weekly for a while [metrics and questionnaires]. Patients are seen for brief med-checks at infrequent intervals. Subjects are assessed using sensitive standardized scales. Patients are asked, "How’s it going?" Lots of differences.
Those are concrete differences between the RCTs that we read about in our journals and the actual practice of medicine. But there’s another glaring difference. The RCTs are done by the company that has the patent on the drug, and who obviously wants to sell it widely for a maximal price. This blog and many others are filled with examples where the RCT is selectively-analyzed/reported to achieve that end. For example, all of the studies of Atypical Antipsychotic Augmentation of Treatment Resistant Depression studies reported in this series were statistically significant, but the Strength of the Effect for the primary outcome variables wasn’t reported. Here are the values extracted in the Spielman et al meta-analysis for those outcomes and the values I got using their same methodology [Cochrane Handbook] in the Geodon® and Rexulti® reports [also showing the IDS-SR Effect Sizes where available]:
| SOURCE | MADRS/HAM-D [d] | IDS-SR [d] | ||
|
|
||||
| Spielmans et al |
0.32 | 0.14 | ||
| Ziprasidone |
0.25 | – | ||
| Brexpiprazole 1mg-3mg |
0.29 | 0.16 | ||
| Brexpiprazole 2mg |
0.29 | 0.20 | ||
The observer rated MADRS/HAM-D numbers are in the weak to moderate range, but the self-rated scales are dramatically near-null – Statistically Significant, but trivial Effect from the Subject’s perspective. So with Data Transparency, willing independent analysts, and cooperative journal editors we could correct a lot of the misbehavior and get all the information.
But these short-term industry run "Approval" RCTs really are still nothing more than a starting place for understanding either efficacy or safety, even at their best. Perhaps the analogy of a model airplane to a Jumbo Jet might be a more reasonable way to conceptualize their place. The real test is when the drugs are put into general use by Clinicians and Patients. And we often don’t hear about those results until well down the line when the side effects and harms start showing up in court-rooms. Patients have plenty of time in waiting rooms getting "screened" or watching tv getting "detailed" by DTC ads, but very little time in the med-check world of Managed Care getting carefully evaluated before or after prescriptions are written. And the time allotted for reporting is… well it just isn’t in the program. That clown at the World of DTC Marketing in the last post [when pigs can fly…] has plenty of suggestions about how we can talk to our patients about the provocative questions raised by his silly ads when they "ask the doctor", but apparently doesn’t know that we already don’t even have time to do an adequate medical interview much less what he suggests.
PharmalotBy Ed SilvermanDecember 30, 2015
In a controversial move, the American Medical Association recently called for a ban on advertising prescription drugs and medical devices directly to consumers. The effort is largely symbolic because any ban would have to be authorized by Congress. But doctors resent the increasing pressure the ads place on them to write prescriptions out of concern patients will switch physicians. And they argue that many ads aimed at consumers promote more expensive medicines. Richard Meyer, a former Eli Lilly marketing executive, who is now an industry consultant who runs the World of DTC Marketing blog, explains why he believes the AMA is misguided.

By Richard Meyer, pharmaceutical consultantLast month, members of the American Medical Association declared that drug makers should stop advertising their products directly to consumers because they feel it contributes to an increase in health care costs and pushes patients to ask for products that either they may not need or is not right for them. This approach is, at best, misguided, and, at worst, ignores the benefits of direct-to-consumer advertising for patients. According to a study on DTC marketing that was conducted by Eli Lilly, 25 percent of patients who were prompted to visit their doctor after seeing an ad were given a new diagnosis. Of those patients, 43 percent were a “high priority” diagnosis for a serious health condition, like diabetes or hypertension.
That same study indicated that 53 percent of physicians felt that DTC ads lead to better discussions with patients because patients are better educated and informed. In addition, a 2004 Food and Drug Administration survey of physicians and patients found that exposure to DTC ads spurred 27 percent of Americans to make an appointment with their doctor to talk about a condition they had not previously discussed. Another study found that the small print in a drug ad was strongly associated with patients contacting their health care providers.
But there is more.
A November 2006 report from the General Accountability Office noted that only 2 percent to 7 percent of patients who requested a drug in response to a drug ad ultimately received a prescription for the medicine. In another study, DTC ads increased the likelihood that a patient would initiate a dialogue with a physician to request an advertised drug. In still another study, which was conducted in 2010 by Prevention magazine, 79 percent of those queried said they sought a specific product. At that point, the magazine had tracked such data for 13 years and the figure was an all-time high. Yet, only 19 percent of the patients actually received the product they sought, an all-time low. DTC advertising increases awareness of health problems and leads to a better informed and educated patient who can engage their physician in a dialogue rather than a monologue.
So what’s really going on here?
First, insurers are taking more prescription writing power away from doctors. They first want patients to try generic medications which now make up 88 percent of all available prescription drugs. Second, higher patient copayments for office visits and insurance mean consumers are “shopping” for health care and health care treatments. This makes doctors very uncomfortable. Even with all these changes, research continually validates the notion that patients view their doctors as the gatekeepers to their prescription medicines. If a doctor doesn’t feel it’s right for the patient, then they won’t write for it.
The AMA would be better served to remind doctors to have the so-called “weight” conversation with patients, since obesity is at epidemic levels here in the United States and is costing health care billions of dollars. Patients should be warned of potential problems, if any, and, in conjunction with insurers, a comprehensive wellness plan should be developed. DTC advertising leads patients to their health care providers and, depending on the health condition, does not lead to high-priced unnecessary scripts. The AMA should reach out and work with pharma to improve DTC marketing, not request a ban on all DTC ads.

Robert Spitzer, the author of the DSM-III, died on Christmas Day at age 83. While the commentaries so far praise his removing psychoanalysis from psychiatry, he gets mixed reviews for the system he developed. All agree that he became the most influential psychiatrist of his time.
 I’ve never been able to say much about him or his DSM-III without hearing back that my opinion is suspect because I am a psychoanalyst. Unlike the modern KOLs, I wouldn’t argue that it’s not a Conflict of Interest that colors what I think. It definitely does. But I agree that psychoanalysis shouldn’t have been a part of psychiatry or psychiatric diagnosis in the first place. The fact that it became allied with psychiatry in America was neither what Freud wanted, nor what happened in the rest of the world. Parenthetically, psychoanalysis as a non-denominational discipline has actually thrived since the separation from psychiatry. But that’s another story.
I’ve never been able to say much about him or his DSM-III without hearing back that my opinion is suspect because I am a psychoanalyst. Unlike the modern KOLs, I wouldn’t argue that it’s not a Conflict of Interest that colors what I think. It definitely does. But I agree that psychoanalysis shouldn’t have been a part of psychiatry or psychiatric diagnosis in the first place. The fact that it became allied with psychiatry in America was neither what Freud wanted, nor what happened in the rest of the world. Parenthetically, psychoanalysis as a non-denominational discipline has actually thrived since the separation from psychiatry. But that’s another story.
At the time of the DSM-III introduction, I thought it read like an academic exercise more focused on itself than the patients it purported to classify. Spitzer wasn’t a clinician, and his DSM-III ablated established categorical clinical distinctions in the service of improving inter-rater reliability, most notably, Melancholic Depression versus depression as a symptom. Presumabely, he also wanted to get rid of depressive neurosis in the process, but that stroke opened the floodgates to the later SSRI/Atypical/TRD craze, and stopped affective disorder research in its tracks for no clear reason. I personally thought the DSM-III subtracted from rather than added to or improved upon, so I paid it little mind. But within a very short time, psychiatry was undergoing a massive sea change, and there was clearly a push towards pharmaceutical treatment and research along with changes in third party payments. And for some of us, it was a time for a change in employment.
 Looking back on those days, I don’t think psychiatry changed just because of Robert Spitzer. Robert Spitzer’s DSM-III was the more public face of a broader effort with strong political and economic undercurrents that transcended the stated scientific agenda. I doubt that Robert Spitzer; or the Medical Director of the APA [Dr. Mel Sabshin]; or those colleagues Spitzer called "the invisible college" [the neoKraepelinians centered at Washington University] had any inkling of the power they were ceding to the pharmaceutical and insurance industries in the process, or how that scenario would play out over the next three decades.
Looking back on those days, I don’t think psychiatry changed just because of Robert Spitzer. Robert Spitzer’s DSM-III was the more public face of a broader effort with strong political and economic undercurrents that transcended the stated scientific agenda. I doubt that Robert Spitzer; or the Medical Director of the APA [Dr. Mel Sabshin]; or those colleagues Spitzer called "the invisible college" [the neoKraepelinians centered at Washington University] had any inkling of the power they were ceding to the pharmaceutical and insurance industries in the process, or how that scenario would play out over the next three decades.
The biggest ringer in the story is what we now call the KOLs, the group of psychiatrists in high places who joined up with PHARMA for personal and institutional gain. I would never have dreamed that would happen. I doubt if Robert Spitzer did either. They’re still there, and without a thorough cleansing, I doubt that there will be any meaningful resolution of anything. You know who they are. Their names are spread throughout the posts on this and many other blogs. Robert Spitzer was as victimized by their antics as the rest of us.
 So back to Spitzer’s legacy. Alongside of his depression gaff, there’s another place where I think he deserves to be personally blamed. He kept a lot of what he was doing under his hat, shared only with his confidants. So the DSM-III process was something of a politically maneuvered bloodless coups d’etat orchestrated in concert with an inner circle of the APA. That behind-the-scenes oligarchy has persisted, undermining any sense that the APA represents its membership [better characterized these days as its following]. Whether the stealth and all the palace intrigue was necessary or not [ends justifying the means], it has persisted as a style for 35 years to all of our detriment.
So back to Spitzer’s legacy. Alongside of his depression gaff, there’s another place where I think he deserves to be personally blamed. He kept a lot of what he was doing under his hat, shared only with his confidants. So the DSM-III process was something of a politically maneuvered bloodless coups d’etat orchestrated in concert with an inner circle of the APA. That behind-the-scenes oligarchy has persisted, undermining any sense that the APA represents its membership [better characterized these days as its following]. Whether the stealth and all the palace intrigue was necessary or not [ends justifying the means], it has persisted as a style for 35 years to all of our detriment.
 When something’s wrong, it’s a lot easier to say what’s wrong than it is to know what to do about it. The cross-fire of the academic-industrial complex and Managed Care is a choke-hold that rarely lets one come up for breath. Here we are about to finally escape from under the patent-life of all these drugs, and up pops Rexulti®, an Abilify® clone, and the FDA grants them a benefit-of-the-doubt approval for this Atypical Antipsychotic Augmentation of Treatment Resistant Depression indication coming right out of the pipeline.
When something’s wrong, it’s a lot easier to say what’s wrong than it is to know what to do about it. The cross-fire of the academic-industrial complex and Managed Care is a choke-hold that rarely lets one come up for breath. Here we are about to finally escape from under the patent-life of all these drugs, and up pops Rexulti®, an Abilify® clone, and the FDA grants them a benefit-of-the-doubt approval for this Atypical Antipsychotic Augmentation of Treatment Resistant Depression indication coming right out of the pipeline.
The only thing I know to do is to try to chase down and make public how much of a cliff-hanger this FDA Approval was, and to try to make the actual state of affairs in the profile of this drug as clear as possible [see a story: the beginning of the end…]. I hardly know what to make of the way this was published – two ghost-written industry prepared articles with only one academic author, a notorious KOL at that, in back to back articles in the same journal with a lot of identical text; attempting to sell the idea that changing a protocol in midstream is an acceptable bit of science; living on p-values while ignoring Effect Size measurements. It’s really a bit too much all around.
If there’s a prime example to illustrate why the Direct-to-Consumer advertisements need to be taken off the air, psychiatric medications are the prime contender. The ads have been relentless, misleading, and the very real damage from the overmedication in part fueled by these ads is all around us. The AMA has recommended that they be banned [damn the torpedoes! full speed ahead…]. I’d up that recommended to demanded were it up to me. Those ads are as destructive as the cigarette ads used to be, and deserve the same fate.
Starting to work in a public clinic after 20 years in practice as a psychotherapist and five years of retirement was something of a shock. Besides the general polypharmacy, the number of people taking Atypical Antipsychotics was staggering to me. At the time, Seroquel® was the number one selling drug in the country, and everyone was on it. I didn’t understand how that had come to be, but I did know what to do about it. So by my second year back at work, no patients in our clinic who were not psychotic were on those drugs.
Recently, they changed the policy at the clinic, and so we’ve taken on a new crop of patients. And here they come again – this time on a greater variety, though primarily on Seroquel® and Abilify®. And this time around, I’ve seen three cases of Tardive Dyskinesia [TD]. Mercifully, two were mild and seem to be slowly clearing, but one isn’t. It’s the patient I wrote about in blitzed… and some truths are self-evident… who was on an outrageous drug regimen. The further along we get with tapering the drugs, the more apparent her TD symptoms – the worst case I’ve personally ever seen. She’s never been psychotic, and her diagnosis is probably best characterized as an attachment disorder.
That’s the case that’s in my mind as I read these Atypical-Antipsychotic-Augmentation-of-Treatment-Resistant-Depression articles, and probably why I’ve stayed glued to this topic for so long. I’ve realized what others may have already known – the Atypical-Antipsychotic-Augmentation-of-Treatment-Resistant-Depression has been advertised far and wide and has given permission to clinicians of many ilks to add Atypicals to their poly-pharmacy regimens. And it’s no surprise that the cases I’ve seen are on Seroquel® and Abilify®. They’re the only two FDA Approved for this indication, and are legally allowed to advertise:

Both Seroquel® and Abilify® are now off patent, so the ads will disappear. But now the FDA has approved Rexulti® for this indication based on these lackluster-at-best clinical trials I’ve been reviewing, so we can count on seeing in Rexulti®-in-Depression ads soon. Even worse, it has gotten that approval at the beginning of its patent life, so it will be with us for a very long time, perpetuating this practice of Atypical-Antipsychotic-Augmentation-of-Treatment-Resistant-Depression. Can the Direct-to-Consumer ads be far behind?
What’s doubly tragic to me, is that as this patient has awakened from her drug-induced-stupor, it’s obvious that she’s a particularly kind of patient well known to me and others in my tribe. She’s a person with an attachment disorder [often called Borderline Personality] whose adulthood has been chaotic because of her difficulty with and in relationships. She’s been seen by a variety of mental health types, and has been obviously chronically overmedicated [with her participation]. Such patients finally wear out trying to make the world adapt to their volatility and childish needs, and become very treatable in later adulthood. This process has been called a Hegira of therapies [Gerald Adler]. So here she is, finally ready to perhaps live a different and more fulfilling life, and she may have developed an unnecessary and disfiguring iatrogenic illness to contend with for the rest of her days.
In-so-far as I can tell from my recent review of the literature, there’s no real treatment for TD except not to get it in the first place. And although there are exceptions [some early onset cases], one’s chance of developing TD on antipsychotics increases steadily the longer you’re on the drugs. Add to that the danger of developing what’s called the Metabolic Syndrome [weight gain and Diabetes] and the risk side of the risk/benefit equation becomes very heavily weighted. Furthermore, these days, people are kept on medicines once started indefinitely. So this approval of Rexulti® for Atypical-Antipsychotic-Augmentation-of-Treatment-Resistant-Depression has the potential to do some real damage – as that done in my patient to absolutely no good purpose. No short term RCT is ever going to make this risk apparent. And no case of TD is worth the minimal effects the drugs might have on depression.
I started the last post with a borrowed literary device, but I was just practicing. In The French Lieutenant’s Woman, John Fowles popped into his story and spoke in the first person with the reader. He did that, or at least claimed he did that, because he couldn’t decide how to end his novel. He ended up writing two endings. Well that’s my problem with this story. I’ve got two different endings, both of which matter to me, but they aim at substantively different conclusions and there’s no resolving them. So I’m just going to have two endings and be done with it. but first…
Thus, with one strong·ly positive trial and supportive evidence from two additional trials, there is adequate evidence of efficacy to approve this product for the adjunctive treatment of MDD.
But is that what strong means? Very significant? We actually have other ways of looking at strong, called strength of effect or Effect Size. I mentioned them in in the land of sometimes[2]… and in the land of sometimes[3]…. The Primary Outcome Variable in these studies is the MADRS score at 6 weeks compared to baseline [a continuous variable]. The usual measure of Effect Size would be Cohen’s d, but they neither calculated it nor gave us enough parameters to do it ourselves. But they did use the MADRS score to tally Responders [≥ 50% reduction] and Remitters [≥ 50% reduction and ≤ 10]. So I looked to see how strong they were in the 2MG study. I found the NNT [Number Needed to Treat] and OR [Odds Ratio] for the original Efficacy Population and the Final Efficacy Protocol using their Table 2 and the Supplement:
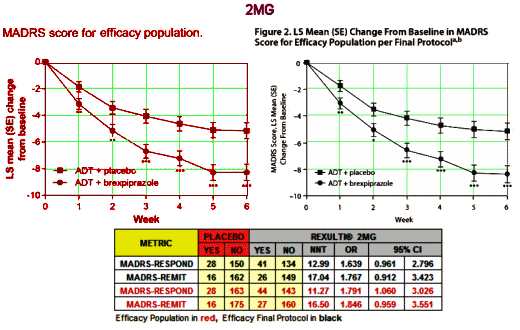
The NNT of 13 says that you have to treat 13 patients to get one that exceeds what you’d get with placebo alone. That’s a lousy number, on the other end of the scale from strong. It’s a weak if any effect. No matter which study you look at, or which population you use, these are all lackluster numbers. And the Odds Ratios consistently under 2.0 are low. Here’s the 1MG AND 3MG study, equally dismal:
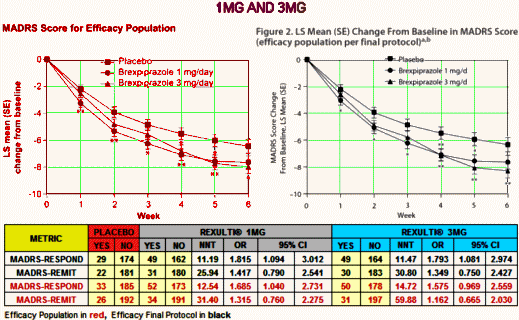
You might notice that the graphs look better than the numbers. You can do that using a change from baseline scale that obscures the fact that the MADRS is a 60 point scale and then using the SEM for limit markers rather than the SD or the 95% CIs which might show how close these lines really are [I think of it as presentation spin].
Since I couldn’t do the Cohen’s d for the MADRS scores, I had a look at the HDRS-17:
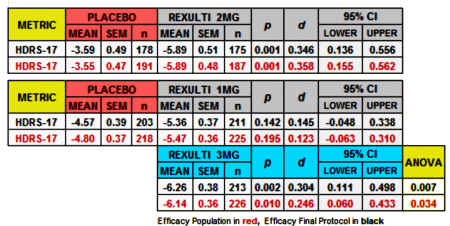
Finally, since I have a spreadsheet that converts the usual Mean, SEM, n figures into Effect Sizes [Cohen’s d AKA Standardized Mean Diffierence], I kept going and put in all of the continuous Secondary Outcome Variables for the two studies [recall d=0.2 is weak, d=0.5 is moderate, and d=0.8 is strong]:
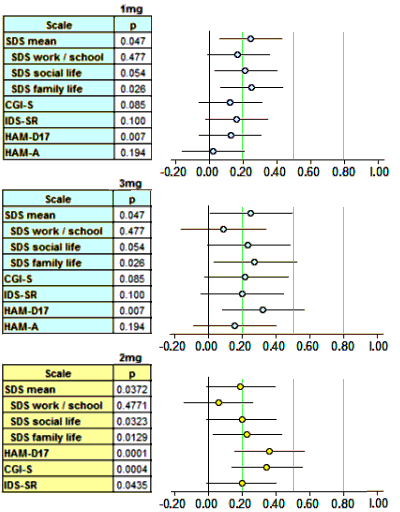
Scanning down the forest plots, there’s nothing at all that I see justifying the strong moniker. Brexpiperazole just didn’t leave many footprints in the sand. And as Spielmans et al pointed out in their meta-analysis [the extra mile…], the self-reported depression scale [IDS-SR] is in the hardly-noticed range. Searching the text of the two articles, measurements of Effect Size are barely mentioned or reported. Certainly they play no important role in their analysis or discussion. So I’ll add that to my list of complaints about these papers.
I think I’ll borrow a literary device from a couple of favorites: T.S. Eliot in East Coker and John Fowles in The French Lieutenant’s Woman. It’s where the author jumps out of his story and speaks in the first person with the reader about his thoughts while he’s writing, then returns to the thread of the narrative…
When I first became belatedly aware of all the corrupted science and the industrial interference in our psychiatric literature, I was outraged and spent my time, like a lot of us, expressing my dissatisfaction, decrying and lamenting the sorry state of affairs. As I learned more, I figured out that a key feature of the problem was the lack of data transparency, and joined in that effort. I’ve been pleased that there has been some incremental movement in that regard from the collective efforts. But there’s something else. While I was pleased to be a part of finally getting the truth published about Paxil Study 329, it was 14 years after the fact when Paxil has been long off-patent [and hopefully mostly forgotten].
 I decided that what we need is part of a long-standing Preventive Medicine principle – Early Detection and Prompt Intervention. We need to jump on the Clinical Trials that are jury-rigged early, and stay on them from the very start before too much damage has been done. So on the first of every month, I scan through the journals and look for Clinical Trials that need vetting and get started. That’s how I got on this topic of Augmentation with Atypical Antidepressants in
I decided that what we need is part of a long-standing Preventive Medicine principle – Early Detection and Prompt Intervention. We need to jump on the Clinical Trials that are jury-rigged early, and stay on them from the very start before too much damage has been done. So on the first of every month, I scan through the journals and look for Clinical Trials that need vetting and get started. That’s how I got on this topic of Augmentation with Atypical Antidepressants in  Treatment Resistant Depression. And I’m following it as far as it will take me. I have no illusion that one old man can change much, but I’m hoping that some younger folks will catch the bug and take up the mantle. We’ll get nowhere if we don’t jump on these things quickly.
Treatment Resistant Depression. And I’m following it as far as it will take me. I have no illusion that one old man can change much, but I’m hoping that some younger folks will catch the bug and take up the mantle. We’ll get nowhere if we don’t jump on these things quickly.
So much for my little interlude coming clean about my agenda. It explains why I’ve taken to sticking in my amateur statistical pieces and why I plan some future looking-up-tips-and-tricks – passing on the tools of vetting I’ve picked up along the way. In fact, I’m about to pass one on right now – the wonders of a sometimes favorite website – Drugs@FDA.com…
When I found out that Brexpiprazole had been approved for the both Schizophrenia and Antidepressant Augmentation, I went straight to the FDA site [Drugs@FDA.com], but it wasn’t there. Sometimes, they lag and I wasn’t surprised. After I’d been through the papers, I’d worked up a good case of outrage, but it occurred to me that on my FDA trip, I’d only looked for brexpiprazole. Usually it’s under both names. But on a lark, I went back and looked under Rexulti®, and there it was! On that site, you look for the Approval History, Letters, Reviews, and Related Documents , then the Review, then the Medical Review(s) [all 238 pages!]. But don’t be discouraged, there’s usually a good table of contents for easy exploring. And there it was on page 95:
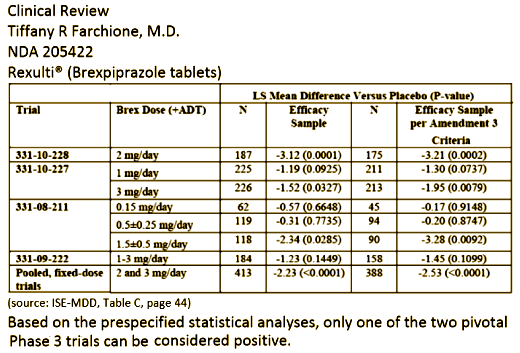
7.4. Adjunctive Treatment of MDD
7.4.1. The Sponsor conducted two adequate and well – controlled trials to asse ss the efficacy of brexpiprazole for the adjunctive treatment of MDD. Based on the prespecified statistical analysis plan, only one of these trials (Study 331-10-228) was positive. The Sponsor acknowledges that Study 331-10-227 was not positive based on the pre-specified plan, but provides a number of arguments to support the concept that brexpiprazole should nonetheless be approved for this indication.
Approval of the adjunctive MDD indication would have to rest on one of two possible scenarios: 1. Study 331-10-227 can be considered a positive trial based on the retrospective application of Amendment 3 criteria and use of the per protocol population instead of the intent – to – treat population for the primary analysis. 2. Study 331-10-228 can be considered “strongly positive” and, thus, approval can be based on this single study. Study 331-10-227 would then be viewed as “supportive evidence” for this approval.
In the first scenario, it is true that efficacy of brexpiprazole 3 mg/day was demonstrated in Stud y 331 – 10 – 227 using the per protocol population. When Amendment 3 was implemented, the data were still blinded. At that time, the Sponsor assured the Division that the primary analysis would still be based on the ITT population and did not request any modification to the statistical analysis plan. Only once the data were unblinded and the “near miss” was discovered did the Sponsor ask us to consider these alternative analyses. There is no question that Amendment 3 resulted in randomization of more appropriate subjects; it is the retrospective application of the amendment criteria to unblinded data that is problematic. This may be a case in which hindsight is 20/20 — had the Sponsor asked to modify the statistical analysis plan to use the per protocol population for the primary efficacy analysis, we may have granted that request. There is precedent for such decisions as long as the blind has not been broken. But, in this case, no such request was made. Changing the statistical analysis plan after the blind has be en broken is not appropriate ; thus, we cannot simply accept this as a positive trial.
On the other hand , the results of Study 331-10-227 can be viewed as supportive evidence, allowing for approval based on the second scenario. As noted above, there is no question that Amendment 3 resulted in randomization of more appropriate subjects, and no question that this would have been a positive study if the Sponsor had used the Amendment 3 criteria from the start of the study. Indeed, if one restricts the analysis of 331-10-227 to only those subjects randomized after Amendment 3 (taking the question of blinded vs. unblinded selection of subjects for analyses off the table), the 3 mg/day brexpiprazole+ADT would be statistically superior to placebo+ADT (p=0.003 3 ).
In addition to these considerations , the Sponsor picked a particularly stringent statistical method for dealing with multiple comparisons — a method which we advised against. If the Sponsor had chosen a less stringent method (for instance, testing 3 mg first then 1 mg), the study would have been positive. The Sponsor also employed the Hochberg procedure in Study 331-08-211. Similar to Study 331-10-227, one dose group (1.5 + 0.5 mg/day) in 331-08-211 yielded a p<0.05 (p=0.0285) on the primary endpoint. But, al so similar to 331-10-227, the pre – specified multiplicity adjustment required a lower threshold. Of note, a retrospective application of Amendment 3 criteria to the study population in 331-08-211 would also make the treatment effect in this dose group statistically superior to placebo (p=0.0092).
One can also reasonably consider Study 331-10-228 a “strongly positive” study — in a population of individuals with a history of multiple failed antidepressant trials and prospectively demonstrated suboptimal respons e to an additional antidepressant, the addition of 2 mg/day of brexpiprazole yielded an average additional symptom improvement of just over 8 points on the MADRS. This was 3 points beyond the improvement in placebo — a difference that was highly statistically significant at p=0.0001. These results are both clinically and statistically meaningful.
Thus, with one strongly positive trial and supportive evidence from two additional trials, there is adequate evidence of efficacy to approve this product for the adjunctive treatment of MDD.
I’m picking up the thread of a story in the middle, because I didn’t realize it was a story with installments until I was well into it. It started with December’s American Journal of Psychiatry that had two Publicly funded Clinical Trials that looked like Infommercials [Experommercials] to me, one being about augmenting antidepressants with Geodon® in the mythologic ‘treatment resistant depression.’ I was upset that it was publicly funded [creative funding I…, creative funding III & some other things…]. Looking at it, I noticed the lead author and the author of the accompanying editorial had done a meta-analysis of ‘augmentation’ in 2009. And so I looked at that meta-analysis which I thought was reasonable, but I disagreed with the conclusion [skepticism unchanged…]. That lead to a more recent [2013] metanalysis that was more extensive, one I agreed with [the extra mile…, a worksheet post…]. Then I ran across a pair of articles on a new Atypical Antipsychotic [brexpiprazole], virtual clones of each other. Brexpiprazole [Rexulti®] was recently approved for augmentation of ‘treatment resistant depression’ [extending the risk…, a postscript…]. That’s a story in my book, though I don’t yet have a title. This post picks up in the middle with those two brexpiprazole articles.
In extending the risk…, I couldn’t stop talking about these two articles being clones. The only difference is that one used two doses [1MG AND 3MG] and the other used only one [2MG]. Why didn’t they just do three doses in one study? We can answer one that right off the bat. The FDA requires two studies for approval, so that’s all there is to say about that. We’re so used to it that we don’t even register that this is a blatant example of a pharmaceutical company using our scientific literature for commercial purposes. They had different ghost-writers from the same firm, but they must’ve been in adjacent cubicles because big pieces of are actually cut and pasted. The Clinical Trials were submitted on the same day. I’m just going to mention a few of their differences. Here are the stats from clinicaltrials.gov:
| STUDY | CLINICAL TRIAL | START | STOP | N | SITES | ~N/SITE | ||||||
|
|
||||||||||||
| 1MG AND 3MG | NCT01360632 | JUN 2011 | SEP 2013 | 677 | 71-92 | 7-10 | ||||||
| 2MG | NCT01360645 | JUL 2011 | MAY 2013 | 379 | 57-59 | ~6 | ||||||
If you look at the SECONDARY OUTCOME VARIABLES, there is a big difference [which I’ll discuss later]. These studies are done by Contract Research Organizations [MGH?], and they have a huge number of SITES all over the world. They do this to speed things up, as any given SITE has to only recruit a small number of subjects [the possibility of introducing error is obviously increased by this ploy]. Here are the papers [full text available]. It’s worth your while to actually scan through them. I’ve named them [1MG AND 3MG and 2MG] for clarity:
1MG AND 3MGby Thase ME, Youakim JM, Skuban A, Hobart M, Zhang P, McQuade RD, Nyilas M, Carson WH, Sanchez R, and Eriksson H.Journal of Clinical Psychiatry. 2015 76[9]:1232-1240.
2MGby Thase ME, Youakim JM, Skuban A, Hobart M, Augustine C, Zhang P, McQuade RD, Carson WH, Nyilas M, Sanchez R, and Eriksson H.Journal of Clinical Psychiatry. 2015 76[9]:1224-31.
Following the prospective treatment phase, patients were eligible for entry into the double-blind randomized treatment phase if they had inadequate prospective ADT response, defined as < 50% reduction in HDRS-17 total score between baseline and end of the prospective phase, with an HDRS-17 total score of ≥ 14 and a Clinical Global Impressions-Improvement scale (CGI-I) score of ≥ 3 at the end of the prospective phase. While this study was ongoing, additional analyses were performed on data from a completed phase 2 study of similar design… It was found that a small number of patients in that study had seemingly adequate improvement in Montgomery-Asberg Depression Rating Scale (MADRS) and CGI-I scores at various times during the prospective treatment period, but subsequent worse scores at time of randomization. These patients did not show a consistent lack of response and would have been considered adequate responders if evaluated at another time point during the prospective phase. A number of these patients showed significant improvement again during the randomized phase, even if continuing on ADT alone.
In order to exclude patients with seemingly variable response to ADT, this study’s protocol was amended in March 2012 during the enrollment phase and prior to database lock to specify that patients had to meet more refined inadequate response criteria throughout prospective treatment (HDRS-17 score ≥14, <50% reduction from baseline in HDRS-17 as well as <50% reduction in MADRS total score between start of prospective treatment and each scheduled visit, and CGI-I score ≥3 at each scheduled visit) to be eligible for randomization. The investigator was also blinded to the revised criteria. Both the protocol amendment and the resulting primary analysis were discussed and agreed with the relevant regulatory authorities (US Food and Drug Administration).
In order to exclude patients with seemingly variable response to ADT, this study’s protocol was amended to specify that patients had to meet more refined inadequate response criteria throughout prospective treatment (a HDRS-17 score ≥14; < 50% reduction from baseline in HDRS-17, as well as <50% reduction in MADRS total score between start of prospective treatment and each scheduled visit, and CGI-I score ≥3 at each scheduled visit) to be eligible for randomization and also to blind the investigator to the revised criteria.
"MADRS score (primary end point). In the efficacy population per final protocol, mean reduction from baseline to week 6 in MADRS total score for brexpiprazole 3 mg showed greater improvement (−8.29) compared with placebo (−6.33; least squares [LS] mean difference = −1.95; 95% CI, −3.39 to −0.51; P = .0079) (Figure 2). Mean change in MADRS total score for brexpiprazole 1 mg was −7.64 versus −6.33 for placebo (LS mean difference = −1.30; 95% CI, −2.73 to 0.13; P = .0737) (Figure 2).""Mean change in MADRS total score for the efficacy population also showed improvement for brexpiprazole 3 mg versus placebo (−1.52; 95% CI, −2.92 to −0.13; P = .0327) but did not reach the level of statistical significance required for multiple comparisons according to the prespecified statistical analysis. The mean improvement for brexpiprazole 1 mg versus placebo was less than that for 3 mg (−1.19; 95% CI, −2.58 to 0.20; P = .0925) (Supplementary eFigure 1)."
"MADRS score (primary end point). Mean reduction from baseline to week 6 in MADRS total score was greater for brexpiprazole compared with placebo (LS mean = −8.36 vs −5.15; LS mean difference = −3.21 [95% CI, −4.87 to −1.54], P = .0002; efficacy population per final protocol) with difference between treatment groups apparent from the first week onward (Figure 2).""Similar results were seen for brexpiprazole versus placebo in the efficacy population (LS mean = −8.27 vs −5.15; LS mean difference = −3.12 [95% CI, −4.70 to −1.54], P = .0001) (Supplementary eFigure 1)."
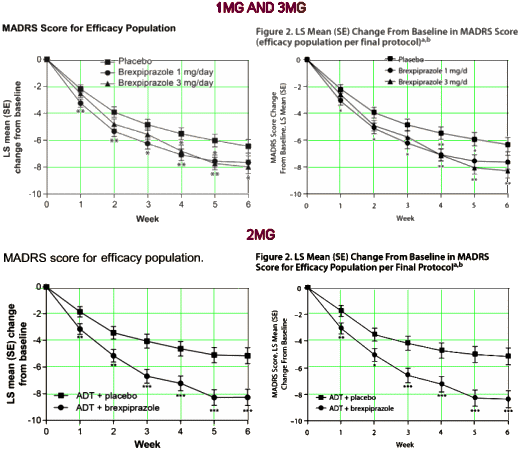
First, in 2MG they are on solid ground with either protocol. But that wasn’t true in 1MG AND 3MG. With the efficacy population, they report a P = .0327 for the 3mg dose in the narrative [above], but after correcting for false-discovery rate with multiple variables with the protocol specified Hochberg correction [too many SECONDARY OUTCOMES], both 1mg and 3mg are insignificant. With the efficacy population per final protocol, the 3mg dose achieves significance. If you look at those upper graphs long enough, you can see the difference – slight, subtle, just enough to pull the numbers into significance [don’t squint]. And remember, they need two positive studies for FDA approval.