

Posted on Wednesday 1 October 2014
The European Medicines Agency (EMA) is set to finalise its new policy on access to clinical trial data this weekOut-Law.com30 Sep 2014Guido Rasi confirmed to the European Parliament that the EMA’s management board will approve the new policy on Thursday, according to a report by EU news website euractiv.com. A spokesperson for the EMA confirmed to Out-Law.com that the body will release details of the new policy after the management board’s meeting either on Thursday afternoon or Friday. "Our new policy will spell out clearly what will be the principles to gain access," Rasi said, according to the euractiv.com report. "The decisions, however, will always be taken by the EMA. Next Thursday, our management board will approve this new version of the policy. That should give us, from the start of 2015, the possibility to start to actively make data available. By 2016, we should have the first clinical trials data available."
Paul Ranson, a specialist in the regulation of clinical trials at Pinsent Masons, the law firm behind Out-Law.com, said that the EMA is expected to announce that it will, as a first step, make Clinical Study Reports (CSRs) available after product assessment subject to the redaction of identified commercially confidential information. CSRs are documents that contain all the details of a clinical trial that are submitted to regulators by a company seeking authorisation to market a new product.
The EMA previously announced that its new policy would not address issues concerning the disclosure of individual patient data (IPD) generated from clinical trials. Ranson said that the EMA is still engaged with stakeholders in discussing what conditions to impose on access to IPD. However, he said many companies have already adopted their own policies and allow restricted access to IPD using a gatekeeper model, which is where a trusted third party control access to the data and impose privacy safeguards. Some, like GSK, have created their own ‘safe havens’ where certain data can be viewed but not extracted, whilst others, such as Bristol Myers Squibb, are making the data available through a collaborative safe haven, Ranson said. "An important consideration for companies is what is said about data reuse and data sharing in the consent form signed by participants," Ranson said. "This is likely to be more of an issue for past trials as data sharing and reuse can be addressed in the consent forms for new trials."
Earlier this year the European Ombudsman Emma O’Reilly criticised EMA draft plans which she said would, if introduced, allow clinical trials data only to be accessible on screen using an interface provided by the EMA. O’Reilly claimed further "wide restrictions" would apply to how the data could be used, something which the EMA denied at the time. It said it intended to make clinical trial data publicly accessible online and that restrictions it was considering were designed to prevent those who access the information using it for commercial gain.
Competing interests surround the issue of whether and to what extent clinical trial data should be made publically available. Many stakeholders believe that medical research would be enhanced if there was full transparency from the results of clinical trials. However, others believe that at least an element of confidentiality is required to protect the significant investments drug companies make in carrying out clinical trials and developing new drugs. They believe rival companies should not be able to piggy-back on their investment and launch new products onto market at their expense.
Expert in life sciences Helen Cline of Pinsent Masons said: "There is a whole spectrum of benefits that could arise from making the clinical trial process and clinical trial data more transparent from improving the efficiency of drug discovery to the public health benefits.” Earlier this year, the Council of Ministers and the European Parliament voted in favour of a new Clinical Trials Regulation which will require pharmaceutical companies and other medical researchers to post results of all their European clinical trials on a publicly-accessible database.
Having just spent the year poring over the information from a real clinical trial, I have a better sense of what’s needed to thoroughly vet such a study. None of it is commercially sensitive in the way I understand the terms. The only way it would be commercially sensitive would be if the truth meant the drug couldn’t be as profitable as the maker might like because it wasn’t very effective or had prohibitive side effects – which are the reasons to do a trial in the first place. How effective is it? What are the harms? So I’m not sure what they mean by Commercially Confidential Information.
For an Efficacy evaluation, one needs the a priori protocol which spells out how the study is to be conducted; the outcome variables and what parameters will be collected; and finally how the data will be analyzed. There’s nothing about commerce in all of that. The Adverse Event analysis requires the rawest form available of the adverse events for each subject – before being transcribed and coded. Again, nothing about commerce in any of that – just the clinical facts.
The CSR [Clinical Study Report [CSR] is a "write up" of the study. If it contains the original protocol and the tables of the raw results for the outcome variables for each subject, it’s fine for the efficacy evaluation. But if it’s simply a summary and only contains the processed or analyzed information, it’s of no use. You can jury rig a CSR just as easily as a paper for publication. So when they throw around the term, CSR, we need to know specifically what’s in it. What’s needed is the protocol and the IPD [individualized participant data]. If they aren’t both there, it’s not data transparency. It’s something else [see it matters…]. So when I read, "new policy would not address issues concerning the disclosure of individual patient data" – I worry. For an evaluation of Adverse Events, one needs the CRF’s [Clinical Report Forms]. We need to know what the evaluator saw and what the subject said – not some compilation or already coded list. I don’t read about that here and so I worry some more.
 I am personally indebted to him for wiping the vasoline off of my own lenses. I hated seeing what he showed us, but am grateful that it finally saw the light of day. Senator Grassley gets loud Kudos for his work in Congress and the moral strength to pursue it, but Paul Thacker was the one who did the leg work and the head work to make it all happen. I’m absolutely sure that he opened a lot more eyes than just mine with his work, and with his later exposures of ghostwriting when he was with POGO. He reminded us of a medical ethic that had gotten lost in a lot of circles along the way. We owe him so much more than he claims for himself…
I am personally indebted to him for wiping the vasoline off of my own lenses. I hated seeing what he showed us, but am grateful that it finally saw the light of day. Senator Grassley gets loud Kudos for his work in Congress and the moral strength to pursue it, but Paul Thacker was the one who did the leg work and the head work to make it all happen. I’m absolutely sure that he opened a lot more eyes than just mine with his work, and with his later exposures of ghostwriting when he was with POGO. He reminded us of a medical ethic that had gotten lost in a lot of circles along the way. We owe him so much more than he claims for himself… 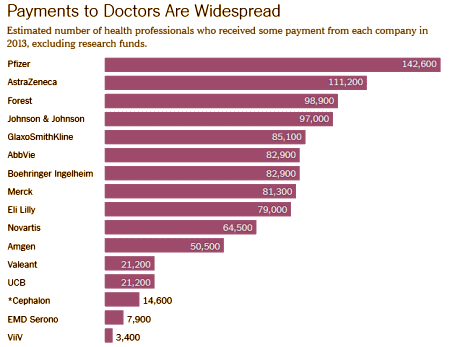
 I declare ongoing resentments against Pfizer on any number of grounds: the way Zoloft® was approved by the FDA was insider trading [see [
I declare ongoing resentments against Pfizer on any number of grounds: the way Zoloft® was approved by the FDA was insider trading [see [

 They concluded:
They concluded:
![Niccolò di Bernardo dei Machiavelli [1469 – 1527]](http://1boringoldman.com/images/machiavelli.gif "Niccolò di Bernardo dei Machiavelli [1469 – 1527]") I’m aware that I tell this story as if it is a Masterpiece Mystery or a Machiavellian court drama, but I come by that quaisi-paranoia honestly. The pharmaceutical industry may have moved on from the psychiatric drug arena, but they’ve left a legacy of disreputable wheelings and dealings that is inescapable. PHARMA doesn’t want Data Transparency, and I would expect them to use every bit of their considerable power and financial resource to undermine it wherever possible. Without the cloak of secrecy surrounding Clinical Trial results, none of the CNS blockbusters of the last few decades would’ve ever left the gate. And that’s likely true as well industry-wide with other drugs.
I’m aware that I tell this story as if it is a Masterpiece Mystery or a Machiavellian court drama, but I come by that quaisi-paranoia honestly. The pharmaceutical industry may have moved on from the psychiatric drug arena, but they’ve left a legacy of disreputable wheelings and dealings that is inescapable. PHARMA doesn’t want Data Transparency, and I would expect them to use every bit of their considerable power and financial resource to undermine it wherever possible. Without the cloak of secrecy surrounding Clinical Trial results, none of the CNS blockbusters of the last few decades would’ve ever left the gate. And that’s likely true as well industry-wide with other drugs.  After months of anticipation, a Chinese court found the GlaxoSmithKline subsidiary in China guilty of bribing doctors, hospital officials and other non-government personnel, and fined the drug maker more than $490 million,
After months of anticipation, a Chinese court found the GlaxoSmithKline subsidiary in China guilty of bribing doctors, hospital officials and other non-government personnel, and fined the drug maker more than $490 million,  Glaxo unit in China, pleaded guilty to bribery-related charges and was given a three-year suspended sentence. There are varying reports, however, whether he will be deported or required to remain during that time. Four other senior Glaxo managers in China also received suspended sentences of between two and four years…
Glaxo unit in China, pleaded guilty to bribery-related charges and was given a three-year suspended sentence. There are varying reports, however, whether he will be deported or required to remain during that time. Four other senior Glaxo managers in China also received suspended sentences of between two and four years…