Institute for Safe Medication Practices
QuarterWatch
Monitoring FDA MedWatch Reports
January 28, 2015
Executive Summary
The U.S. Food and Drug Administration’s Adverse Event Reporting System [FAERS] — based on MedWatch reports – is the government’s primary safety surveillance system designed to identify harms from therapeutic drugs. For the last six years these vital data have formed the core of ISMP’s QuarterWatch™ drug safety reports. In this issue we decided to look closely at the system itself. Our conclusion: it seems clear that this drug safety monitoring system is in need of modernization. It suffers from a flood of low quality reports from drug manufacturers and has not yet been updated for the changing environment in which drugs are marketed to health professionals and consumers. We discuss key problems below and offer some recommendations as an organization that relies heavily on data collected through FAERS. This issue of QuarterWatch includes two recently released calendar quarters of FAERS data, from 2013 Q4, and 2014 Q1. To provide a broader perspective, the main analysis focuses on the 12 months ending with 2014 Q1, and includes all adverse event reports received by the FDA in that one-year period. Previous issues of QuarterWatch have focused on a subset of these case reports, those with a serious outcome and reported by patients in the United States…
The recent announcement that the NIH and FDA intend to beef up clinicaltrials.gov and insist that it be used as intended has been welcomed news and we’re eager to see how their resolve is implemented [see ]. But this is equally, if not more important. The original charge of the FDA was
SAFETY – Adverse Events cum Serious Adverse Events in modern parlance. And that’s as it should be. An individual doctor can’t possibly discover adversity until it happens, and the incidence of dangerous toxicity is often low enough to pop up late in a practitioner’s use of the drug. Clozoril is a good example. It’s a hell of a fine antipsychotic, the best there is. I’ve only seen it used a few times in desperately ill cases that were unresponsive to any usual measures, and it lived up to expectations in all of the cases where I’ve seen. But one case of fatal agranulocytosis is enough to remove it from a doctor’s personal formulary. Fortunately, the frequent monitoring of blood counts seems to give ample warning for

preventive discontinuation, but the possibility of an ominous outcome is still always in mind when the drug is used. There are innumerable examples throughout medicine where infrequent cataclysmic toxicity is what rules drug usage.
EFFICACY was an FDA add-on on the 1960s after Thalidomide, an irony in that Thalidomide is, by report, one of the most
effective antiemetics around and is still used where there’s no chance of fetal exposure. In summary, there’s no possibility of a risk/benefit ratio without both a numerator [
SAFETY] and a denominator [
EFFICACY]. And an accurate risk/benefit ratio is part of every prescription written, whether it’s conscious or not. It’s part of the medical
auto-pilot, or at least it should be.
One of the important dimensions of the risk/benefit equation is TIME. If you are rendered totally miserable by a case of poison ivy, I can radically [and safely] improve your lot with a short, but generous tapering course of corticosteroids – the powerful suppressor of inflammation. But I’ve got to be sure you don’t have a condition like a Herpes eye infection that can be exacerbated by even a short course. The same with an asthma attack that doesn’t respond to bronchodilators. You might say time is on your side, because the diseases themselves are time-limited. But if you show up with a generalized case of Rheumatoid Arthritis, the corticosteroids are a double edged sword. The disease is going to be around for a while – long enough for the prodigious down side of chronic steroid treatment to raise its ugliest of heads. So the job is to look for a long term treatment from Day 1, and only use steroids up front, if at all. The same thing is true of anti-anxiety drugs or narcotic analgesics. Benzodiazepines are the cat’s meow for a crisis state with unmanageable anxiety, but danger, danger for the patient with a lifelong anxiety disorder because of tolerance and addiction ["if one is good, two is better"]. The same goes for narcotics. Just right for a heart attack or a kidney stone, but danger, danger, danger for some chronic back and other pain problems. Once you know they work, it’s harder and harder not to take them, and then you enter the nightmare of addiction.
And even if our short term Clinical Trials are conducted and analyzed thoroughly and honestly, they tell us little about long term use. That’s a real problem for psychoactive drugs [all of them]. It’s also a problem for many treatments of medical conditions that stick around for a long time [or forever]. So we need to know about the long term toxicity, and our Clinical Trials can’t tell us – emphasis on can’t. So we need a long term monitoring system to give us the information we need, and we just don’t have it. The current FDA MedWatch Report System just doesn’t seem to be it. Good for them that they noticed.
Besides a reporting system that works, identification of adverse effects is often a problem in and of itself, particularly in a poly-medicated world. You can’t report a side effect to a drug if you don”t know it’s a side effect. I was once called to see a patient in the hospital to be worked up for a neuromuscular problem with periodic extreme weakness. A morning muscle biopsy hae been aborted in the face of him having a panic attack. He was a fit male lying in the bed with a wet towel on his forehead in an obvious terror state – very hard to interview. He reported episodes of extreme weakness, mostly at work, worsening over several months. He worked in in a factory that made electrical cables, the big kind that are strung from tower to tower across the landscape. He ran the last machine in the process that wrapped the twisted cable onto huge spools for storage and transport. It was all done in a big un-airconditioned warehouse that was like an oven in the Georgia summers. The punchline was, "Doc. I feel like if I could just sweat, I’d be all right!" Flash back to a routine physical the winter before when he had mild hypertension and was started on medications. After trying several different drugs he couldn’t tolerate, he saw a specialist and was put on a new drug, Inderal, brand new at the time. It’s a Norepinephrine Beta-Blocker that incidentally turns off the sweating process. So he was right. And his panic disorder? As a boy, he had witnessed the slow demise of a favored uncle from Lou Gehig’s Disease, which is what he privately thought he had. His Blood Pressure? "White Coat Syndrome." It was normal when taken by his wife at home. Once we knew what was wrong, the cure was simple. Another example close at hand is the frequent misinterpretation of antidepressant withdrawal symptoms as a recrudescence of the original depression or the emergence of an anxiety syndrome, and the problem medication is restarted. Unfortunately, that maneuver works and perpetuates the cause. So side effects, adverse events, are difficult to spot.
In a world where pharmaceutical companies minimize adverse effects, third party payers pay for drive-by doctors’ visits, and beautiful people parade across our television screens during the mumbled warnings, collecting adverse events with an accurate numerator, denominator, and time-marker is no trivial task.
DavidHealy.org has modeled a shot with his
Rxisk, but it, too, has a voluntary IN and OUT and lacks official conduits to the Agencies that matter. But it seems to me that the FDA has fallen down on this part of their mandate. I think a place to start a pilot project would be adding a pharmacy prescription-based system, periodically querying ongoing prescriptions and those that go unrefilled for selected medications. But this is not close to my area of expertise. I know we need to make it both easy and desirable for doctors and patients to report their adverse experiences without adding to the CYA or silly screening burden of doctor’s visits.
The last time I had my blood drawn, I was asked if I’d heard voices telling me to hurt myself or others in the last three weeks [among other things]. I was tempted to say, "No, it’s been well over a month."
A doctor’s medical auto-pilot will hold a lot of information, but it needs to be programmed. The task ahead on this problem is a big [and important] job for the FDA. It’s what CME [Continuing Medical Education] was originally designed to pass on…


 heavily colored by a quest to find a marketable pick-the-antidepressant test. It has played out among the usual suspects of entrepreneurial psychiatry in high places, spread through the pages of academic peer-reviewed journals, and been nurtured in the departments of psychiatry at some
heavily colored by a quest to find a marketable pick-the-antidepressant test. It has played out among the usual suspects of entrepreneurial psychiatry in high places, spread through the pages of academic peer-reviewed journals, and been nurtured in the departments of psychiatry at some 
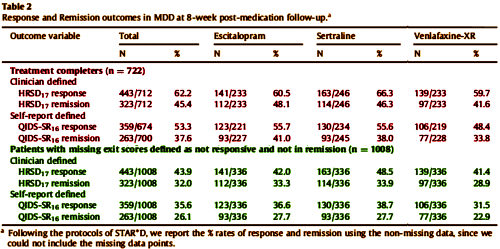
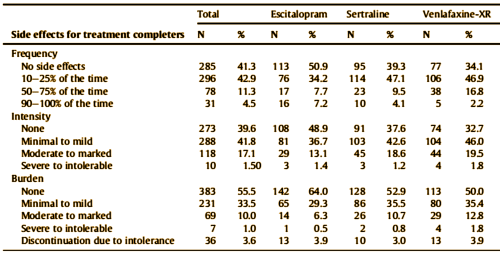
 So here we are three decades after the first SSRI, two decades beyond the hypothesis that there’s some special way these drugs can be used to increase their efficacy looking at a study that says that they’re all the same. And we know what’s coming – a string of studies sifting through all tests neuro looking for something that will point to one or the other drug [with no real rationale that such a test might exist]. We have here a study financed by a commercial firm aiming for a future commercial product, authored by its employees, and published in a journal edited by some of the authors who are also involved with the company. The ticket into a peer reviewed medical journal is presumably the academic credentials of some of those authors.
So here we are three decades after the first SSRI, two decades beyond the hypothesis that there’s some special way these drugs can be used to increase their efficacy looking at a study that says that they’re all the same. And we know what’s coming – a string of studies sifting through all tests neuro looking for something that will point to one or the other drug [with no real rationale that such a test might exist]. We have here a study financed by a commercial firm aiming for a future commercial product, authored by its employees, and published in a journal edited by some of the authors who are also involved with the company. The ticket into a peer reviewed medical journal is presumably the academic credentials of some of those authors.
 My first thumbing through the DSM-III in 1980 landed me in Major Depressive Disorder [MDD], and it’s where I’ve remained. I’ve written about it enough here to thoroughly earn my 1boringoldman moniker [see
My first thumbing through the DSM-III in 1980 landed me in Major Depressive Disorder [MDD], and it’s where I’ve remained. I’ve written about it enough here to thoroughly earn my 1boringoldman moniker [see  Obviously what we want is the raw data from the Clinical Trial – the same thing that the pharmaceutical company has to work with. In the good old days, only one agency was involved. Nowadays, the study sites [CRC – Clinical Research Centers] are coordinated and managed by a Clinical Research Organization who collects the data and passes it on to the PHARMA. The CRO/PHARMA analyze it and pass the summary to the writers. Once it’s drafted, the guest authors come into the picture and send it to a journal. If there is an FDA NDA submission, it comes from the PHARMA, prepared by some mixture of the CRO, writers, and PHARMA. What we see on the FDA web site is the FDA’s Reviewer report about the submission, but not the contents of the submission.
Obviously what we want is the raw data from the Clinical Trial – the same thing that the pharmaceutical company has to work with. In the good old days, only one agency was involved. Nowadays, the study sites [CRC – Clinical Research Centers] are coordinated and managed by a Clinical Research Organization who collects the data and passes it on to the PHARMA. The CRO/PHARMA analyze it and pass the summary to the writers. Once it’s drafted, the guest authors come into the picture and send it to a journal. If there is an FDA NDA submission, it comes from the PHARMA, prepared by some mixture of the CRO, writers, and PHARMA. What we see on the FDA web site is the FDA’s Reviewer report about the submission, but not the contents of the submission. 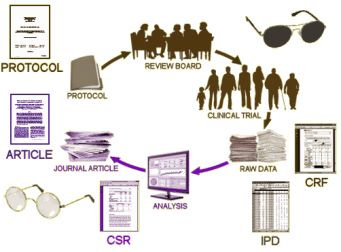
 HUNTSVILLE, Ala. [WHNT] – Alabama Psychiatric Services confirms it will end services on February 13, 2015. It cites a decrease in funding from Blue Cross/Blue Shield and a change in how the insurance company covers behavorial health services as the reason for the closure. An employee at the Madison facility confirmed the closure earlier today, as did several patients. The Madison employee tells us the closure of APS’ statewide offices could impact as many as 200,000 patients. In north Alabama, APS also has facilities in Cullman, Decatur and Florence. The Madison location is on Lanier Road off Hughes Road. The company posted this message on
HUNTSVILLE, Ala. [WHNT] – Alabama Psychiatric Services confirms it will end services on February 13, 2015. It cites a decrease in funding from Blue Cross/Blue Shield and a change in how the insurance company covers behavorial health services as the reason for the closure. An employee at the Madison facility confirmed the closure earlier today, as did several patients. The Madison employee tells us the closure of APS’ statewide offices could impact as many as 200,000 patients. In north Alabama, APS also has facilities in Cullman, Decatur and Florence. The Madison location is on Lanier Road off Hughes Road. The company posted this message on